Definition
Fæokromocytomer er tumorer opstået i de kromaffine celler i binyremarven, som producerer katecholaminerne adrenalin, noradrenalin og dopamin. Lignende tumorer opstået i de sympatiske ganglier kaldes paragangliomer 1.
Hvad omfatter denne NBV?
Denne NBV omhandler fæokromocytom og sympatisk paragangliom (herefter blot benævnt paragangliom).
Hvad omfatter denne NBV ikke?
Denne NBV omfatter ikke tumorer opstået i de parasympatiske ganglier (parasympatiske paragangliomer også kaldet ”head and neck tumors”).
Diagnosekoder (ICD)
- DD350A Fæokromocytom
- DE275 Øget katecholaminsekretion
- DD356B Godartet tumor i paraganglion
- DC741 Kræft i binyremarv
- DC755B Kræft i andet paraganglion
Visitation
Initial biokemisk diagnostik kan foregå på afdeling med hovedfunktionsniveau, yderligere diagnostik af fæokromocytom og paragangliom er en medicinsk endokrinologisk regionsfunktion. Patienter med dissemineret sygdom henvises til specialiseret onkologisk afdeling og afdeling med højt specialiseret endokrinologisk funktion.
Forekomst
Fæokromocytom og paragangliom er sjældne lidelser. Den årlige incidensrate i Danmark af fæokromocytom er tidligere rapporteret at være 1,9 per million2. Incidensen er dog stigende til 4,7 per million3 på grund af udredning af binyreincidentalomer og familiescreening ved de genetiske former.
- Adrenale: 85 – 90 %
- Bilaterale: 5 – 10 %
- Maligne: 10 %
- Del af en arvelig sygdom: 40 %
Ætiologi
40 % skyldes nedarvede ændringer i regulatoriske proteiner (se afsnit om genetik).
Symptomer og kliniske fund (4)
- Hypertension (anfaldsvis eller persisterende) 81 %
- Hovedpine 60 %
- Palpitationer 59 %
- Anfaldsvise symptomer 58 %
- Svedtendens 52 %
Andre symptomer: Angst, bleghed under anfald, tremor, brystsmerter, dyspnø, kvalme, vægttab, svimmelhed, udmattelse og flushing.
Blandt patienter som undersøges for muligt fæokromocytom og paragangliom forekommer svedtendens, palpitationer, bleghed, tremor og kvalme hyppigere hos de patienter, som får bekræftet diagnosen5.
Ubehandlet ses forøget kardiovaskulær morbiditet og mortalitet6. Forværring af symptomer, og evt hypertensiv krise (hos den ubehandlede) kan udløses af lægemidler (dopamin D2 receptor antagonister, β -blokkere, opioider, tricykliske antidepressiva og serotonin- og noradrenalinoptagshæmmere (SSRI/SNRI)), indledning af anæstesi og ved manipulation af tumor.
Udredning (Figur 1)
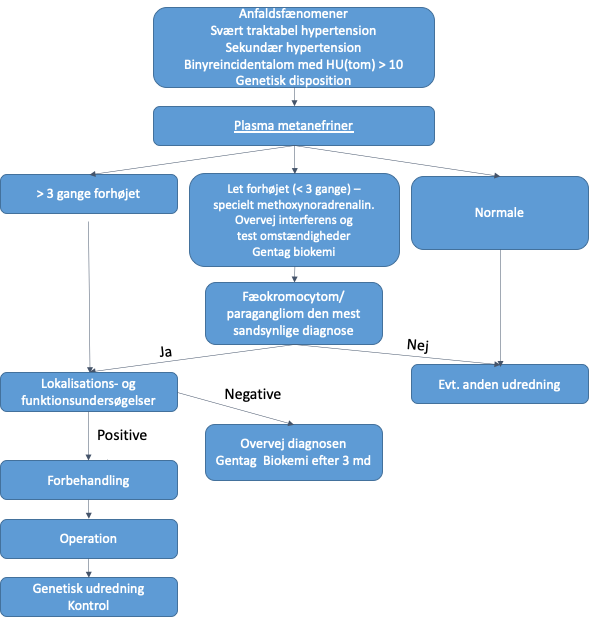
Indikation for udredning
- Anfaldsfænomener
- Anfaldsvis og svært traktabel hypertension
- Mistanke om s(NBV sekundær hypertension)
- Heterogent binyreincidentalom
- Binyreincidentalom med Hounsfield Unit (HU) > 10på CT skanning uden kontrast) (NBV Binyreincidentalom)
- Genetisk disposition
Biokemisk diagnostik
Den biokemiske diagnose stilles ved påvisning af forhøjede koncentrationeraf metanefriner (methoxyadrenalin og methoxynoradrenalin), som er inaktive nedbrydningsprodukter fra katecholaminerne i plasma.
| Plasma-metanefriner Analysemetode | Sensitiv analyse som f.eks. liquid chromatography tandem mass spectrometry (LC-MS/MS) |
| Patientforberedelse | Optimale forhold: 1. Undgå koffeinholdige drikke fra aftenen før 2. 30 minutters liggende hvile inden prøvetagning 3. Afholde sig fra fysisk aktivitet forud for prøvetagning (fx cykle eller trappegang) 4. 2 ugers pause med lægemidler, der påvirker analysen (se tabel 1) Af pragmatiske årsager vil man dog ofte tage prøve uden nogen forberedelse og kun gentage prøven under optimale forhold, hvis den første prøve er forhøjet. |
| Tolkning (se figur 1) | Normale plasma metanefrinkoncentrationer udelukker med stor sandsynlighed fæokromocytom og paragangliom, da alle påvirkende faktorer vil øge niveauet af metanefrinerne. Ved lettere forhøjet specielt methoxynoradrenalin (<3 gange øverste normalgrænse) kan falsk positivt testresultat ikke udelukkes. Ny test under optimale forhold overvejes. Isoleret mindre methoxynoradrenalin forhøjelse kan skyldes alder (>65 år), og der kan korrigeres derfor7. Forhøjelse af både methoxyadrenalin og methyxonoradrenalin er sjældent et falsk positivt fund. Nyresyge (stadie 3-4) har ca. 50-100% højere metanefriner end nyreraske. |
Tabel 1: Læge- og nydelsesmidler der påvirker p-metanefriner
| p-methoxynoradrenalin | p-methoxyadrenalin | |
| Tricykliske antidepressiva | ++ | – |
| α-blokkere (non-selektive) | ++ | – |
| α-blokkere (alpha1-selektive) | – | – |
| β-blokkere | + | + |
| Calcium antagonister | – | – |
| Monoamine oxidase inhibitorer | ++ | ++ |
| Sympatomimetika | ++ | ++ |
| Methyldopa | ++ | – |
| SNRI | ++ | – |
| Paracetamol (assay afhængigt / interferens med assay) | ++
| – |
| Stimulerende stoffer – Koffein, kokain, nikotin | ++ | + |
Øvrige biokemiske analyser
Chromogranin A frigives fra neuroendokrine celler, men ikke specifikt fra fæokromocytom eller paragangliom. Måling af chromogranin A vanskeliggøres af flere interagerende faktorer (hyppigst protonpumpe hæmmer behandling og nedsat nyrefunktion), som giver ophav til falsk forhøjede værdier. Chromogranin A anvendes derfor ikke diagnostisk, men forhøjede værdier kan benyttes ved monitorering af sygdomsforløbet.
Måling af noradrenalin og adrenalin i døgnurin er muligt men sjældent indiceret.
Tumorlokalisation
Patienten udredes med lokalisations- og funktionsundersøgelser. Billeddiagnostik afventes til diagnosen er biokemisk bekræftet.
Lokalisationsundersøgelse
CT skanning er første valg – bemærk at en binyretumor med HU ≤ 10 ved skanning uden kontrast for alle praktiske udelukker et fæokromocytom8.
MR scanning udføres hvis CT scanning er kontraindiceret.
Funktionsundersøgelser (tabel 2)
Tabel 2 (modificeret efter 9)
| Første valg | Andet valg | Tredje valg | |
| Sporadisk fæokromocytom | 18F-FDOPA
| 68Ga-DOTA-peptid | 18F-FDG |
| Paragangliom Metastatisk sygdom SDH varianter | 68Ga-DOTA-peptid | 18F-FDOPA eller 18F-FDG | |
| Arveligt fæokromocytom (ikke SDH varianter) | 18F-FDOPA | 68Ga-DOTA-peptid | 18F-FDG |
Forkortelser 18F-FDOPA: Fluorin-18-mærket fluoro-l-dihydroxyphenylalanin; 68Ga-DOTA-peptid: Dækker over Gallium-68-mærkede gængse somatostatinanaloger som er i brug; 18F-FDG: Fluorin-18-mærket fluorodeoxyglucose
68Ga-DOTA-peptid, 18F-FDOPA og 18F-FDG undersøgelserne laves som positron emissions tomografi (PET)/CT scanninger, således vil lokalisations- og funktionsundersøgelse med disse tracere udføres samtidigt.
Da udbuddet af funktionsundersøgelser i høj grad afgøres af lokale forhold, og da sygdommens lokalisation (fæokromocytom, paragangliom eller metastatisk sygdom) og arvelighed ofte ikke kendes ved behov for lokalisations- og funktionsundersøgelse betragtes 68Ga-DOTA-peptid og 18F-FDOPA PET/CT som ligeværdige første valg i denne situation. Dog kan bemærkes at 18F-FDOPA PET/CT er bedst til påvisning af fæokromocytom pga lav fysiologisk optagelse i binyrerne (i modsætning til 68Ga-DOTA-peptid), mens 68Ga-DOTA-peptider synes bedst til påvisning af ekstraadrenal sygdom.
I den kliniske hverdag benyttes Jod-123-mærket meta-iodobenzylguanidin
(123I-MIBG) SPECT med aftagende hyppighed (dårligere billedkvalitet, længere ventetid på produktion af tracer, undersøgelse kræver patient-fremmøde 2 dage i træk, medikamentelle interaktioner).
Biopsi fra binyretumor er kontraindiceret medmindre fæokromocytom er udelukket.
Behandling
Behandlingen af fæokromocytom og paragangliom er operativ fjernelse af tumoren efter medicinsk forbehandling.
Ved tegn på metastatisk sygdom skal patienten konfereres med specialiseret onkologisk afdeling jvf. visitationsafsnit.
Medicinsk behandling før operation
- Der er international konsensus om ALTID at give medicinsk forbehandling til patienter med fæokromocytom og paragangliom, da induktion af anæstesi, intubation, installation af pneumoperitoneum samt manipulation af fæokromocytomet under operation eller biopsi giver risiko for hypersekretion af katecholaminer med ledsagende hypertensive komplikationer.
- Et randomiseret kontrolleret studie10 fandt ingen forskel på den irreversible noncompetitive α1+2-receptor antagonist phenoxybenzamin (Dibenyline) og den competitive α1– receptor antagonist doxazosin (Carduran Retard) i varigheden af blodtryk udenfor målområde under operation; men dog større hæmodynamisk stabilitet under operationen ved phenoxybenzamin. Baseret på dette samt større klinisk erfaring er phenoxybenzamin første valg til medicinsk forbehandling.
- Øvrig antihypertensiv behandling seponeres/nedtrappes om muligt ved opstart af phenoxybenzamin.
- Primært bør behandling med β-blokker seponeres for at undgå hypertensive anfald betinget af katecholaminstimulation af α-receptorerne uden mulighed for ”rescue” β2-receptor medieret vasodilatation. Diuretika bør primært seponeres, idet fæokromocytom/paragangliom-patienten er volumendepleteret.
Optitrering af phenoxybenzamin
- Forbehandlingen startes ca. 2 – 3 uger før operationen eller tidligere ved betydende symptomer og kan evt. iværksættes under indlæggelse.
- Ved start på α -blokade bør patienten være velhydreret og sengeliggende/hvilende ½ time efter tabletindtag.
- Der anbefales saltholdig diæt.
- Startdosis: f.eks. 10 mg x 1 (til natten) eller 10 mg x 2 med gradvis dosisøgning bestemt af symptomer, siddende og stående blodtryk og puls til behandlingsmål nås (jvf. nedenfor).
- Ved reflekstakykardi (grundet hæmning af negativt feedback via blokade af de præsynaptiske α2-receptorer) med puls > 100 kan opstartes supplerende propranolol eller labetalol behandling – dog tidligst efter 2 dages phenoxybenzaminbehandling.
- Ved intolerable bivirkninger til phenoxybenzamin (typisk svimmelhed og tilstoppet næse) kan doxazosin
- Patienterne accepterer dog oftest bivirkningerne p.gr af den korte behandlingsvarighed.
– Sidste dosis phenoxybenzamin og β -blokker gives aftenen før operation.
Vejledende behandlingsmål:
- Anfaldsfrihed
- Blodtryk siddende < 130/80 mmHg
- Blodtryk stående > 90/45 mmHg
- Puls < 80
Operativ behandling
Præmedicin er altid et benzodiazepinspræparat og ikke f.eks. morfin.
Laparoskopisk adrenalektomi er standard behandling. Ved tumorstørrelse over 8 – 10 cm vil åben operation oftest være nødvendig. Hos patienter, der allerede er unilateralt adrenalektomerede, har bilateral sygdom, eller grundet genetisk disposition er i betydelig risiko for udvikling heraf, kan barkbesparende operation overvejes11. Ved paragangliomer vil den operative procedure afhænge af tumorlokalisation.
Oftest benyttes 2-3 arbejdsporte. Den fridissekerede binyre/tumor lægges i pose (endobag) og trækkes ud gennem en af arbejdsportene. Det er kun sjældent nødvendigt at anlægge dræn peroperativt.
Pt. vil oftest være kirurgisk færdigbehandlet efter et døgn. Pt. kan forvente ømhed i operationsområdet og ømhed/smerter i skuldrene (som skyldes efterladt CO2 i abdomen).
Ved åben kirurgi vil patienten forventeligt være kirurgisk færdigbehandlet efter 3-4 dage. Der vil være restriktioner mht. tunge løft de første 3 uger efter operation.
Anæstesiologiske aspekter
Velgennemført forbehandling inklusiv reetableret væske volumen forhindrer ikke anæstesiologiske komplikationer hos patienter med fæokromocytom og paragangliom, hvorfor operation kun bør udføres på specialiserede afdelinger med erfaring i avanceret kardiovaskulær monitorering og behandling.
Hæmodynamisk instabilitet kan forekomme i forbindelse med kirurgisk og anæstesiologisk stress, manipulation af tumor og veneclamping. Det fordrer en høj grad af samarbejde mellem kirurg og anæstesiolog.
Den hæmodynamiske kontrol over kredsløbet håndteres primært ved anvendelse af magnesiumsulfatbolus og adenosininfusion, af og til i høje doser. Der benyttes vasoaktive stoffer med kort halveringstid, hvorimod længere varende beta-blokade sjældent er nødvendig.
Postoperativt har patienterne, også ved ukompliceret anæstesi og kirurgi, behov for udvidet monitorering og observation i operationsdøgnet. Der kan være fortsat behov for kredsløbsstabiliserende stoffer (eksempelvis ved persisterende phenoxybenzamin virkning), samt endokrinologisk overvågning særlig mhp. hypoglykæmi og binyrebarkinsufficiens. Den postoperative observation bør derfor foregå i intensivt regi.
Opfølgning
Efter komplet resektion er recidivraten i gennemsnit lav (ca. 5% pr 5 års opfølgning)12 – højere ved paragangliomer og patogene genetiske varianter end ved fæokromocytomer. Vedvarende hypertension ses hos op mod 50 % af patienterne.
Formålet med postoperativ opfølgning er:
- at sikre, at patienten er helbredt for sygdommen
- at sikre, at sygdommen ikke recidiverer
- tage stilling til genetisk udredning (se nedenfor)
Plan for ambulant opfølgning
Umiddelbart efter operation
2- 4 uger postoperativt foretages status for fæokromocytomet/paragangliomet med måling af plasma metanefriner samt evt. måling af chromogranin A (hvis forhøjet initialt). Formålet er at sikre, at patienten er biokemisk helbredt.
Efterfølgende årlige kontroller
- klinisk vurdering (specielt måling af blodtryk og vurdering af evt. symptomer)
- kontrol blodprøver
- billeddiagnostik ved forhøjede biokemiske markører (ved præoperativ ukendt markørstatus eller ikke-secernerende fæokromocytom/paragangliom da billeddiagnostik af thorax og abdomen hvert 1-2 år (f.eks MR for at reducere stråledosis).
- Patienter som har fået fjernet begge binyrer substitueres med hydrokortison og florinef (Se NBV primær og sekundær binyrebarkinsufficiens).
Patienter over 40 år ved diagnose med mindre fæokromocytomer (< 4 cm) uden patogen genetisk variant følges i 10 år; øvrige patienter kontrolleres livslangt13.
10 og 20 % af henholdsvis fæokromocytomer og paragangliomer er metastatiske. Metastatisk potentiale kan ikke afgøres ved histologisk undersøgelse af primær tumor, men baseres udelukkende på påviste metastaser. Recidiv er hyppigere blandt patienter med ekstraadrenal sygdom samt ved visse patogenegenetiske varianter (succinatdehydrogenase B (SDHB) og neurofibromatose type 1 (NF1)). Ved påvist metastatisk sygdom er 5 års overlevelsesraten 50 %. Der findes ingen kurativ behandling.
Behandling af metastatisk sygdom sker i samarbejde mellem højt specialiseret endokrinologisk og onkologisk afdeling. Den onkologiske behandling kan omfatte radionuklid behandling (somatostatin receptor agonister eller 131I MIBG terapi), kemoterapi med cyclofosfamid, vincristin og dacarbazin,protein kinase hæmmere samt strålebehandling . Hos asymptomatiske patienter synes en ”wait-and-see” strategi undertiden acceptabel14.
Genetisk udredning af fæokromocytomer
Hyppigheden af arveligt fæokromocytom anslås at være helt op til 40 %.
Hvem bør screenes
Alle patienter bør tilbydes genetisk udredning; dog under hensyntagen til at prædiktorerne for genetisk forandring er:
- Familieanamnese
- Alder < 50 år
- Paragangliom
- Flere fæokromocytomer
- Metastatisk sygdom
På nuværende tidspunkt er der beskrevet mere end 25 gener med arvelig disposition til
fæokromocytom/paragangliom. Dog er der sparsom dokumentation for en række af de nyeste kandidatgener.
Arvelige fæokromocytomer forekommer hyppigst ved følgende tilstande:
| Gen | Klinisk Fænotype |
| RET | MEN-type 2a: medullært thyroidea karcinom og primær hyperparathyreoidisme. MEN-type 2b: medullært thyroideakarcinom og neurinomer. Risikoen for udvikling af fæokromocytom er næsten 50 % ved MEN-2, men fæokromocytomerne er oftest godartede (Se NBV MEN) |
| VHL | Von Hippel Lindau: Retinale angiomer, CNS hæmangioblastomer, pancreas neuroendokrine tumores. 20 % af patienterne med von Hippel Lindau udvikler fæokromocytom. Fra 5 års alder anbefales årlig screening med p-metanefriner og fra 15 års alder også Chromogranin A. |
| NF1 | Morbus Recklinghausen neurofibromatose (type 1): Multiple cutane neurofibromer og café-au-lait pletter, aksiale fregner og Lisch noduli i iris Op til 10 % af patienterne med neurofibromatose udvikler fæokromocytom. Debutalderen er lidt senere end ved andre arvelige former. Der henvises til Sundhedsstyrelsens retningslinjer. |
| SDHA SDHB SDHD SDHC | Arvelige paragangliomer er knyttet til patogene genetiske varianter i det mitokondrielle enzym succinatdehydrogenase (SDH), en komponent af respirationskædens kompleks II. Patogene varianter i enzymets fire subenheder (SDHA, SDHB, SDHD og sjældnere SDHC) er associeret med arvelig fæokromocytom og paragangliom. Debut i ung alder, multiple fæokromocytomer eller paragangliomer og ekstraadrenal tumorer herunder renalcelle carcinom, gastrointestinale stromale tumorer (GIST) og hypofysetumorer kan ses patogene SDHx-varianter. Fæokromocytomer hos patienter med SDHB har ofte malignt potentiale |
| SDHAF2, MAX og THEM127 | Ved patogene genetiske varianter i disse gener ses alene fæochromocytom eller paragangliom |
Der henvises til separate forløbsbeskrivelser inden for de enkelte sygdomme.
Der er publiceret flere algoritmer, som ud fra fx patientens diagnose (fæokromocytom eller paragangliom), debutalder og type af hormon-oversekretion (dopamin, adrenalin, noradrenalin) kan benyttes til at forudse den mest sandsynlige genetiske mutation (se tabel 3).
Tabel 3. Hyppigste genetiske tilstande associeret med fæokromocytom og paragangliom
| Syndrom | MEN2 | VHL | NF1 | PGL1 | PGL5 | PGL3 | PGL4 |
| Gen | RET | VHL | NF1 | SDHD | SDHA | SDHC | SDHB |
| Diagnose alder | 30-40 | 15-40 | 40-50 | 30-40 | 20-40 | ||
| Adrenalt fæo | +++ | ++ | +++ | -/+ | ++ | ++ | |
| Bilateral fæo | +++ | +++ | + | -/+ | -/+ | -/+ | |
| Ekstradrenalt paragangliom | -/+ | + | -/+ | + | ++ | + | +++ |
| Hoved/hals paragangliom | – | -/+ | – | +++ | – | ++ | + |
| Biokemisk profil | A | NA | NA, D | NA, D | |||
| Malignitet | – | -/+ | ++ | -/+ | -/+ | -/+ | +++ |
| Familie anamnese | +++ | ++ | + | ++ | + | + | + |
A: adrenerg, NA: noradrenerg, D: dopaminerg.
Alle syndromerne har dominant arvegang, dvs. 50% gentagelsessandsynlighed for at 1.gradsslægtninge har arvet samme patogene genetiske variant men mange af tilstandene har nedsat penetrans, dvs. ikke alle udvikler sygdom, eller variabel ekspressivitet, dvs. tilstanden kan præsentere sig forskelligt i forhold til symptomer og debutalder.
Genetiske analyser er blevet mere tilgængelige, og af praktiske hensyn vil man i dag ofte foretage samtidig genetisk screening af de hyppigste gener (”gen-pakke”) i stedet for successiv screening. Resultat af genetisk screening vil i fremtiden formentlig bidrage til klassifikation af patienten risiko for metastasering16.
Ved positiv familie anamnese screenes primært genet for den mistænkte sygdom.
Betydningen af sjældent forekommende patogene varianter i EGLN1, EGLN2, KIF1B, IDH1, HIF2A, MDH2, FH, SLC25A11 og DNMT3A og hvem, der bør tilbydes screening, er endnu ikke afklaret.
Betydning af positivt mutations fund
Afhængig af lokalt set-up henvises patienter med patogene genetiske varianter til genetisk rådgivning og/ eller førstegradsslægtninge tilbydes genetisk udredning. Ved spørgsmål omkring genetiske analyser og resultater kan den lokale klinisk genetiske afdeling kontaktes for drøftelse. Påviste MEN mutationer henvises jvf. sundhedsstyrelsens specialeplan til højt specialiseret endokrinologisk afdeling.
Followup kan være hyppigere (fx halvårligt) hos patienter med påvist patogene genetiske varianter (f.eks. SDHB) pga. øget risiko for metastatisk sygdom.
Tovholder
Forfattere
Mai Arlien-Søborg
Andreas Ladefoged Ebbehøj
Ebbe Eldrup
Dorte Glintborg
Pernille Holmager
Per Løgstrup Poulsen
Esben Søndergaard
Claus Larsen Feltoft
Anja Lisbeth Frederiksen (klinisk genetik)
Bidrag fra andre specialer
Per Søndergaard Holt (urolog)
Thomas Kistrorp (anæstesiolog)
Birgitte Ruhnau (anæstesiolog)
Seneste revision: Oktober 2020
Næste revision: Oktober 2023
Referencer
- Lam AK. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocrine pathology 2017;28:213-27.
- Andersen GS, Toftdahl DB, Lund JO, Strandgaard S, Nielsen PE. The incidence rate of phaeochromocytoma and Conn’s syndrome in Denmark, 1977-1981. J Hum Hypertens 1988;2:187-9.
- Ebbehøj AL, Søndergaard E, Trolle C, Stochholm K, Poulsen PL. The epidemiology of pheochromocytoma: increasing incidence and changing clinical presentation. A population-based retrospective study 1977-2015. Endocrine Abstracts (2017) 49.
- Soltani A, Pourian M, Davani BM. Does this patient have Pheochromocytoma? a systematic review of clinical signs and symptoms. Journal of diabetes and metabolic disorders 2015;15:6.
- Geroula A, Deutschbein T, Langton K, et al. Pheochromocytoma and paraganglioma: clinical feature-based disease probability in relation to catecholamine biochemistry and reason for disease suspicion. European journal of endocrinology 2019;181:409-20.
- Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab 2013;98:1100-6.
- Eisenhofer G, Lattke P, Herberg M, et al. Reference intervals for plasma free metanephrines with an age adjustment for normetanephrine for optimized laboratory testing of phaeochromocytoma. Ann Clin Biochem 2013;50:62-9.
- Buitenwerf E, Korteweg T, Visser A, et al. Unenhanced CT imaging is highly sensitive to exclude pheochromocytoma: a multicenter study. European journal of endocrinology 2018;178:431-7.
- Taïeb D, Hicks RJ, Hindié E, et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. European journal of nuclear medicine and molecular imaging 2019;46:2112-37.
- Buitenwerf E, Osinga TE, Timmers H, et al. Efficacy of α-Blockers on Hemodynamic Control during Pheochromocytoma Resection: A Randomized Controlled Trial. The Journal of clinical endocrinology and metabolism 2020;105.
- Castinetti F, Taieb D, Henry JF, et al. MANAGEMENT OF ENDOCRINE DISEASE: Outcome of adrenal sparing surgery in heritable pheochromocytoma. Eur J Endocrinol 2016;174:R9-18.
- Amar L, Lussey-Lepoutre C, Lenders JW, Djadi-Prat J, Plouin PF, Steichen O. MANAGEMENT OF ENDOCRINE DISEASE: Recurrence or new tumors after complete resection of pheochromocytomas and paragangliomas: a systematic review and meta-analysis. European journal of endocrinology 2016;175:R135-45.
- Plouin PF, Amar L, Dekkers OM, et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016;174:G1-G10.
- Hescot S, Leboulleux S, Amar L, et al. One-year progression-free survival of therapy-naive patients with malignant pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2013;98:4006-12.
- Neumann HPH, Young WF, Jr., Eng C. Pheochromocytoma and Paraganglioma. The New England journal of medicine 2019;381:552-65.
- Lenders JWM, Kerstens MN, Amar L, et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: a position statement and consensus of the Working Group on endocrine hypertension of the European society of hypertension. Journal of hypertension 2020.