Forkortelser anvendt i denne NBV
ACC adrenokortikalt karcinom;
ACTH adrenokortikotropt hormon
BNVK binyrevenekaterisation
CBG kortisolbindende globulin
CRH corticotropin releasing hormone
CS Cushings syndrom
CT computer tomografi
DST Dexamethason suppressionstest
FDG flour deoxy glukose
HDDST højdosis Dexamethason suppressionstest
HU Hounsfield Unit
LDDST lavdosis Dexamethason suppressionstest
MR magnetisk resonans
ONDST overnight Dexamethason suppressionstest
PET positron emissions tomografi
UFC døgnurin for frit kortisol
Hvad omfatter denne NBV
Udredning, behandling og opfølgning af endogent udløst Cushings syndrom (CS).
Hvad omfatter denne NBV ikke
Denne NBV omfatter ikke iatrogent udløst CS eller autonom kortisolsekretion (subklinisk Cushing).
Diagnosekoder (ICD)
DE 249 CS UNS
DE 240 Hypofysært betinget Cushings sygdom
DE243 Ektopisk adrenokortikotropt hormon (ACTH) syndrom
DE 248 Anden form for CS
Definition
Klinisk tilstand fremkaldt af kronisk endogen hyperkortisolisme.
Forekomst
Den årlige incidens for Cushings syndrom (CS) (ikke maligne tilfælde) er jvf. et dansk nationalt registerstudie (1) 2,3 cases pr. million indbyggere pr. år. Heraf er incidensen for hhv. hypofysært CS (Mb Cushing /Cushings sygdom) og benigne kortisolproducerende binyreadenomer 1,2-1,7 cases pr. million indbyggere og 0,6 cases pr. million indbyggere pr. år. Bemærk at disse tal er fra perioden før binyreincidentalomer systematisk blev undersøgt for kortisoloverskud (se NBV Binyreincidentalomer).
Ætiologi
Tabel 1:
| Inddeling af Cushings syndrom (2) | Fordeling (%) | Alder (år) | Kvinde/mand |
| ACTH-afhængigt | 70-80 | ||
| Hypofysær (Mb.) Cushing | 60-70 | ||
| – Kortikotropeadenomer | 60-70 | 30-50 | 3-5:1 |
| – Kortikotrop hyperplasi | <1 | ||
| Ektopisk ACTH produktion | 5-10 | ||
| – Maligne neuroendokrine tumorer | 4 | 50-70 | 0.6-1:1 |
| – Benigne neuroendokrine tumorer | 6 | 30-50 | |
| – Okkulte neuroendokrine tumorer | 2 | ||
| IACTH- uafhængigt | 20-30 | ||
| Unilateral adrenal | |||
| – Adenom | 10-22 | 40-60 | 4-8:1 |
| – Carcinom | 5-7 | 10-20, 50-70 | 1.5-3:1 |
| Bilateral adrenal | 1-2 | ||
| – Bilateral makronodulær adrenal hyperplasi | <2 | 50-70 | |
| – Bilateral mikronodulær adrenal hyperplasi | <2 |
Ektopiske ACTH-producerende tumorer er bl.a. lokaliseret til thyroidea (medullær thyroideacancer), lunge, thymus, gastrointestinalkanalen og binyremarv.
Symptomer
- Nedsat muskelstyrke og evt. led- og muskelsmerter
- Vægtøgning, især med trunkal fedme
- Utilpashed
- Depression, mani eller psykoser
- Kognitive problemer
- Nedsat libido
- Oligomenoré/amenoré
- Insomni
(Se ref (3) for yderligere)
Kliniske fund
- Pletorisk udseende med moon face, acne og hirsutisme
- Overvægt med abdominal fedme, buffalo hump, tynde ekstremiteter og proksimal myopati
- Tynd og skrøbelig hud (blåviolette striae, ekkymoser/hæmatomer)
- Hypokaliæmi
- Øget forekomst af type 2 diabetes, hypertension, osteoporose, tromboemboliske episoder og infektioner
- Ektopisk CS udviser typisk et mere udtalt og hurtigt indsættende symptombillede
(se ref (3, 4) for yderligere)
Hvem skal udredes på mistanke om CS?
- Patient med symptomer eller kliniske tegn forenelige med CS og hos personer med flere af de kendte følgesygdomme med en atypisk klinisk præsentation (fx debut i ung alder)
- Binyreincidentalom (link NBV Binyreincidentalomer) og tegn på CS
- Mistanke om androgenproducerende tumor i binyrerne
(se ref (2, 3) for yderligere)
Udredning
Måling af kortisol
Kortisol kan måles i plasma (p), urin og spyt. Spyt-kortisol anvendes ikke rutinemæssigt i diagnostikken af CS i Danmark. Kortisolmålinger er assay-specifikke, så viden om det anvendte assay er nødvendigt.
Etablering af diagnosen
Screeningstests og lokalisation.
Der anbefales nedenstående tests, som kan vælges under hensyntagen til lokale forhold samt forhold hos patienten:
- A) Døgnurin for frit kortisol (Urine free cortisol (UFC))
Der udføres 2-3 konsekutive døgnurinopsamlinger. Forhøjet UFC i mindst én opsamling støtter mistanken om CS. Analysen er upåvirket af øget koncentration af kortisolbindende globulin (CBG). Nedsat nyrefunktion (eGFR <30 ml/min/1,73m2) kan give falsk nedsatte værdier og polyuri (>3 l/dag) falsk forhøjede.
- B) Dexamethason suppressionstests (DST)
Enten som:
Overnight Dexamethason suppressionstest (ONDST), hvor 1 mg Dexamethason indtages kl. 23.00. P-kortisol tages næste morgen (kl. 08-09.00). Normal-værdi <50 nmol/l.
Eller som:
48 timers 2 mg Dexamethason/dag test (lavdosis Dexamethason suppressionstest (LDDST)), hvor 0,5 mg Dexamethason indtages hver 6. time i 2 døgn med måling af p-kortisol 6 timer efter sidste dosis. Normalværdi <50 nmol/l. Undertiden måles også UFC i andet døgn.
Det er uafklaret, om der er forskel på ONDST og LDDST mhp. diagnostik af CS. Undertiden beskrives LDDST som havende højere specificitet og bruges således som bekræftende test.
Ved lægemidler, der påvirker CBG eller metaboliseringen af Dexamethason, kan testene ikke anvendes (3).
- C) Midnats p-kortisol
Testen kræver indlæggelse i 2 døgn for at reducere stress og bør kun udføres på endokrinologiske afdelinger med i rutine i denne. Der anlægges perifert venekateter (hvor på blodprøver tages) i dagstiden,. Kl. 24.00 tages p-kortisol på den sovende patient (5-10 min opvågning før test accepteres). Normal-værdi < 50 nmol/ og gråzoneværdier 50-138 nmol/l (4, 5). Hos en vågen patient (>5-10 min) synes p-kortisol <207 nmol/l at udelukke CS (6). Ved lægemidler, der påvirker CBG, kan testen ikke anvendes (3).
TOLKNING
Diagnosen stilles som udgangspunkt ved to abnorme testresultater udført ved forskellige metoder. Hos patienter med lav prætest sandsynlighed for CS (f.eks. osteoporose hos en 30-årig uden andre stigmata) kan diagnosen dog udelukkes ved én normal test.
CS med enten normal UFC eller normal DST er meget sjældent (men beskrevet) og bør give anledning til genovervejelse af diagnosen og yderligere testning.
Sjældent er tillige cyklisk svingende patologiske kortisolværdier (cyklisk CS), hvor gentagne UFC-målinger må udføres hos patienten i symptomatiske perioder.
Ved kun marginalt forhøjede værdier i plasma og urin (typisk UFC ≤ x 3 forhøjet) må fysiologisk hyperkortisolisme (Pseudo-Cushing) udelukkes. Pseudo-Cushing kan ses ved bl.a. fedme, psykiatriske sygdomme, alkoholmisbrug, dårligt reguleret diabetes samt psykisk og fysisk stress.
Det kan til tider være svært at skelne mellem Pseudo-Cushing og ACTH-afhængigt (specielt hypofysært) CS, og der findes ingen sikker test til dette, men Dexamethason-corticotropin releasing hormone (CRH)-testen (måling af p-kortisol 15 min efter injektion af CRH forudgået af LDDST), hvor p-kortisol >87 nmol/l synes at udelukke Pseudo-Cushing (7), kan undertiden anvendes. I tvivlstilfælde anbefales gentagen testning efter en potentiel udløsende faktor er søgt elimineret. I sidste ende kan watchful waiting være indiceret, idet CS i de fleste tilfælde vil progrediere klinisk og paraklinisk.
Figur 1. Diagnostisk algoritme ved klinisk mistanke om CS.
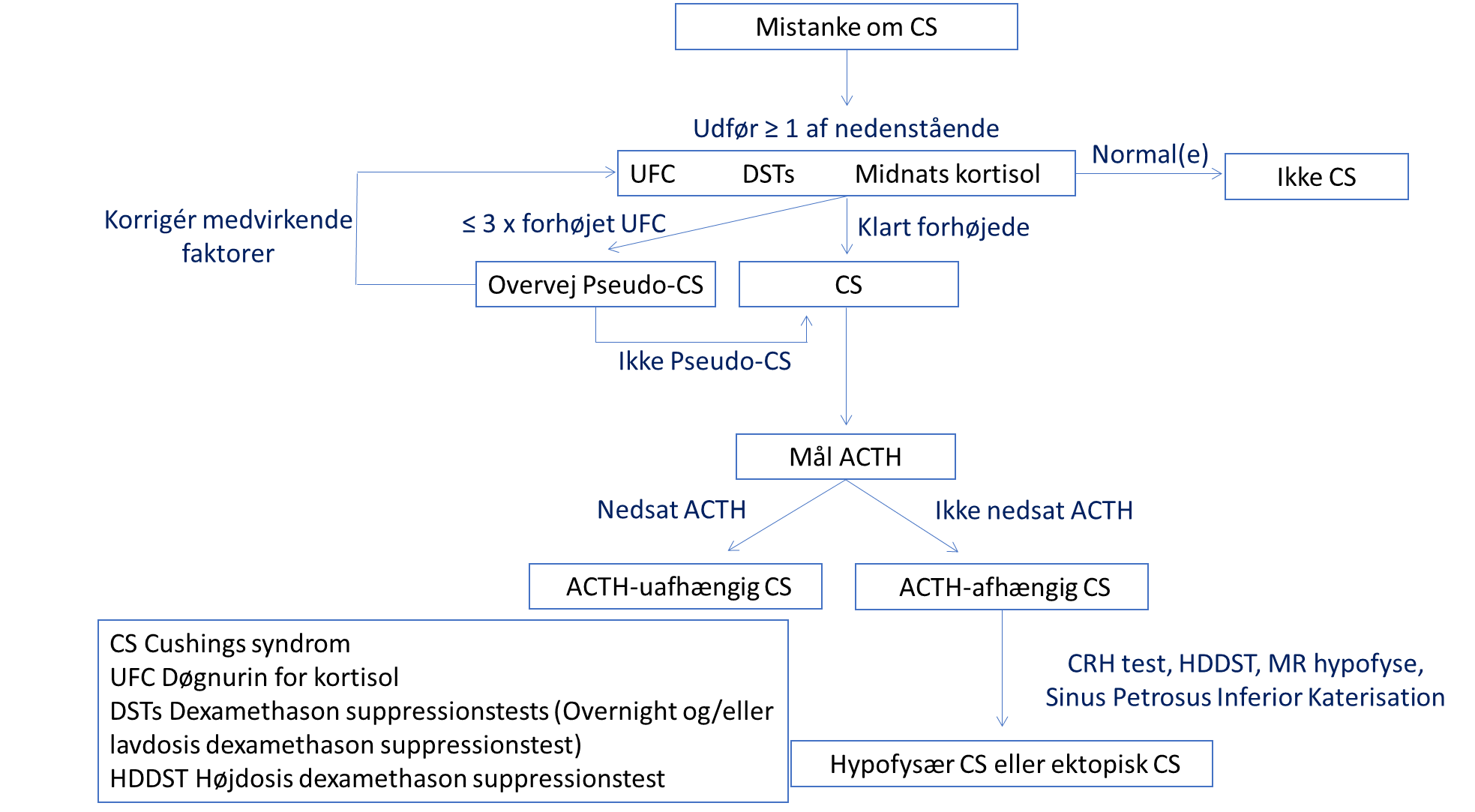
Lokalisations- og billeddiagnostik
Når diagnosen er stillet fortsættes med lokalisationsdiagnostik.
Bestemmelse af p-ACTH
Blodprøven tages kl. 08-09.00.
Ved ACTH-uafhængigt CS ses p-ACTH under assayets nederste normalgrænse, og der kan fortsættes til billeddiagnostik af binyrerne. Ved ACTH-afhængigt CS findes ikke nedsat p-ACTH. Ved ACTH lavt i normalområde gentages ACTH måling (hvor den laveste værdi anvendes) (8) alternativt perifer CRH-test (se nedenfor).
ACTH-uafhængigt CS
Der udføres computer tomografi (CT)-scanning af binyrer. Ved unilateral homogen tumor med Hounsfield Unit (HU) ved tomscanning ≤10 henvises til adrenalektomi (se behandling). Ved unilateral tumor med malignitetssuspekte radiologiske karakteristika udføres CT-scanning af thorax og abdomen (eller flour deoxy glukose (FDG) positron emissions tomografi (PET)/CT-scanning) for at udelukke metastaser fra et evt. adrenokortikalt karcinom (ACC) før behandling. Ved bilaterale tumores laves evt. binyrevenekaterisation (BNVK) med måling af kortisol mhp på evt. unilateral kortisoloverproduktion (9, 10).
ACTH-afhængigt CS
Differentieringen mellem hypofysært CS og ektopisk CS kan være vanskelig. Sandsynligheden for hypofysært CS hos patienter med ACTH-afhængigt CS er ca. 85% (se tabel 1). P-ACTH og p-kortisol er sædvanligvis højere, fænotypen mere udtalt og progression hurtigere ved ektopisk CS end ved hypofysært CS.
Perifer CRH-test
Testene kan være vejledende i ventetiden på sinus petrosus inferior katerisation (se nedenfor) eller hvis mistanken om ektopisk CS er høj.
Ved den perifere CRH-test måles hos den fastende patient p-ACTH og p-kortisol til tiden -15 og 0 min, samt 15, 30, 45 og 60 min efter intravenøs bolus administration af 100 mg CRH. En positiv test med stigning i p-ACTH på ≥ 50% og/eller i p-kortisol på ≥20-30 % har høj
(95 %) positiv prædiktiv værdi for hypofysært CS frem for ektopisk CS; mens en negativ test ikke udelukker hypofysært CS (negativ prædiktiv værdi 70 %). Hvis testen anvendes til at differentiere mellem ACTH-uafhængigt og ACTH-afhængigt CS vil en negativ test pege i retning af ACTH-uafhængigt CS.
Højdosis DST (HDDST)
Ved HDDST gives Dexamethason 2,0 mg p.o. hver 6. time i 2 døgn (i alt 8 doser). I en modificeret udgave indtages 8 mg Dexamethason som engangsdosis kl. 23. Ved begge tests gælder at suppression af p-kortisol (målt 6-8 timer efter sidste tabletindtag) på >50% af udgangsværdien peger i retning af hypofysært CS. Under 2 døgns-HDDST måles også undertiden UFC og >90 % suppression (således relativ værdi) af basal UFC tyder på hypofysært CS. Der er dog betydende overlap i responset mellem hypofysært og ektopisk CS.
2 døgns-HDDST kan laves i direkte forlængelse af LDDST med samme tolkning for begge som beskrevet.
Sinus petrosus inferior katerisation
Sinus petrosus inferior katerisation er guldstandarden til differentiering mellem hypofysært CS og ektopisk CS og kræver en rutineret interventionsradiolog. Sinus petrosus inferior katerisation foregår ved anlæggelse af kateter i sinus petrosus inferior og simultan udtagelse af prøver fra sinus petrosus og perifer vene til bestemmelse af ACTH før og efter CRH-stimulation. En gradient i ACTH på over 2 før og over 3 efter CRH tyder på hypofysært CS. Lateralisering af sinus petrosus ACTH er et usikkert mål forsidelokalisation af den hypofysære sygdom.
Magnetisk resonans (MR)-scanning af hypofysen uden og med gadoliniumkontrast
De fleste ACTH-producerende adenomer er mikroadenomer (< 10 mm), udviser et hypointenst signal og typisk ingen kontrastopladning. I ca. 30-50 % af tilfælde af hypofysært CS kan der ikke påvises et adenom ved MR. Fortolkningen vanskeliggøres af, at der hos ca.
10 % af raske kan påvises et hypofysært incidentalom – dog sjældent større end 6 mm. Derfor kan MR lejlighedsvist anvendes i differentieringen mellem hypofysært CS og ektopisk CS, idet en større tumor (> 6 mm) og en positiv perifer CRH-test vil pege i retning af hypofysært CS.
Øvrig billeddiagnostik og biokemi
Ved stor klinisk og paraklinisk mistanke om ektopisk CS vil billeddiagnostisk undersøgelse af hals, thorax og abdomen ofte accelerere diagnostikken.
CT-scanning er sædvanligvis første valg. Effektiviteten af FDG og gallium-/kobbermærket DOTA-somatostatin analog PET/CT er uafklaret, men specielt sidstnævnte synes lovende. Måling af tumormarkører fraset calcitonin og metanefriner synes ikke at hjælpe med lokalisationen af tumor. På trods af grundige undersøgelser forbliver ca. 20% af de ektopiske tumorer okkulte.
Prognose
Den vigtigste prognostiske faktor hos patienter med benigne hypofyse- eller binyreadenomer er biokemisk sygdomskontrol. Mortaliteten hos patienter med remission adskiller sig således ikke fra baggrundsbefolkningen. Mortaliteten ved ukontrolleret CS er ca. 5 gange forøget.
Hypofysært CS: Postoperativt efter transsfenoidal operation ses remission hos 45-76 % (hyppigereved mikro- end makroadenomer). Trods umiddelbar remission ses recidiv hos 20-36 % (hyppigere ved makro- end mikroadenomer). Recidiv kan opstå mange år efter initial remission.
Ektopisk CS: Remission afhænger af årsag, tumor størrelse og lokalisation. Såfremt der ikke er tale om metastatisk sygdom kan opnås postoperativ remission hos omkring 75 %. Ved recidiv efter resektion af ektopisk ACTH-secernerende tumor bør mistænkes metastatisk sygdom.
ACTH-uafhængigt CS: Ved unilateral benign sygdom (histologisk typisk adenom) kan forventes 100 % remission. ACC har en uhyre dårlig overlevelsesprognose.
Behandlinsmål
- Biokemisk remission/sygdomskontrol med henblik på reduktion af morbiditet og mortalitet
- Behandling af adrenal/hypofysær insufficiens
- Behandling af komorbiditet
Remission defineres som morgen p-kortisol < 138 nmol/l målt få dage efter operation eller UFC lavt i normalområdet < 7 dage efter kirurgisk tumorresektion.
Behandling
Den primære behandling af såvel ACTH-afhængigt og ACTH-uafhængigt CS er kirurgisk. Behandlingen af CS er en specialistopgave (12).
Operativ behandling
ACTH-afhængigt CS
Hypofysær CS:Operativ fjernelse af tumor
Ektopiskt CS: Operativ fjernelse af tumor +/- lymfeknuderesektion; ved tegn på dissemineret malign sygdom da onkologisk behandling
ACTH-uafhængigt CS
Ved unilateral sygdom – unilateral adrenalektomi
Ved bilateral sygdom – bilateral adrenalektomi eller medicinsk behandling. Ved betydende størrelsesforskel på tumorerne og tegn på unilateral kortisol overproduktion ved BVNK da kun unilateral adrenalektomi
Per- og postoperativ behandling
Hvis komplet resektion af tumor er mulig anvendes sædvanligvis peroperativ steroidparaply og postoperativ glukokortikoid -substitution. Det gør sig særligt gældende i forbindelse med adrenalektomi. Profylaktisk steroidparaply og glukokortikoid-substitution kan udelades ved hypofyseoperation, hvis der er adgang til hurtigt svar på p-kortisol (bør måles op til to gange dagligt de første to postoperative døgn samt ved tegn på binyrbarkinsufficiens) og plejepersonalet er veluddannet i observation af patienter med potentiel akut binyrebarkinsufficiens. Fordelen herved er, at man hurtigt (typisk inden for det første operationsdøgn) erfarer om remission er opnået eller ej.
Efter operation for hypofysært CS kan ses forbigående diabetes insipidus efterfulgt af forbigående hyponatriæmi grundet SIADH på ca. 7. postoperative dag (for behandling af øvrig hypofyseinsufficiens henvises til NBV Hypofyseinsufficiens).
Andre behandlinger
Hypofysært CS
Yderligere behandlingsmodaliteter omfatter medicinsk behandling (behandlingsalgoritmen vil være afhængig af sygdommens sværhedsgrad samt patientens køn og komorbiditet og besluttes således på individuel basis), strålebehandling mod hypofyselejet, eller bilateral adrenalektomi.
Medicinsk behandling
Kan anvendes ved svigt af kirurgisamt præoperativt for at kontrollere en betydende hyperkortisolisme.
Adrenale steroidgenese inhibitorer:
- Ketoconazol, startdosis 200 mg x 2-3, max dosis 1.600 mg.
- Metyrapon, startdosis 250 mg x 3-4, max dosis 6.000 mg.
- Osilodrostat startdosis 2 mg x 2, max dosis 60 mg.
- Mitotane, startdosis 500 mg x 3, max dosis 8.000 mg.
- Etomidat, iv. rescue behandling.
ACTH sekretions inhibitorer:
- Cabergolin, startdosis 1 mg pr. uge, maxdosis 7 mg pr. uge.
- Pasireotid, 600-900 mikrog. sc. x 2 dagligt eller 10-40 mg im hver 4. uge
Glukokortikoid receptor antagonist:
- Mifepristone, 300-1.200 mg
Kombinationsbehandling med Ketoconazol og Metyrapon (alle former for CS) samt Ketoconazol, Cabergolin og Pasireotid (hypofysært CS) er en mulighed.
Der kan enten vælges “block and replace” behandling, hvor det endogene p-kortisol reduceres til et minimum (4-6 plasma kortisol målinger i løbet af døgnet under 50-100 nmol/l) og suppleres med hydrokortison i substitutionsdoser. Alternativt en normaliseringsstrategi, hvor der tilstræbes eukortisolisme (4-6 p-kortisol målinger i løbet af døgnet mellem 150-300 nmol/l). Monitoreres endvidere med UFC (under hydrokortison pause) som gerne skal normaliseres.
Stereotaktisk strålebehandling
Kan anvendes ved svigt af kirurgi, samt ved betydende masse effekt eller invasion af det kortikotrope adenom. Stråleterapien gives i fraktionerede doser. Der er indikation for medicinsk behandling indtil, der er opnået effekt af strålebehandling.
Bilateral adrenalektomi
Anvendes ved svigt af øvrige behandlinger i stedet for livslang medicinsk behandling og som rescue behandling ved svær ukontrollabel sygdom. Kan således være indiceret i tilfælde af ex. livstruende komplikationer så som infektioner, lungeemboli, kardiovaskulære komplikationer eller akut psykose. Postoperativt er der risiko for udvikling af Nelsons syndrom (ACTH stigning og tumorvækst). Behov for livslang hydrokortison- og mineralokortikoidsubstitution.
Ektopiskt CS:
Medicinsk behandling
Kan være indiceret ved ikke-sanerbare ektopiske ACTH producerende tumorer – se Hypofysært CS.
Bilateral adrenalektomi
Anvendes ved okkult eller metastatisk sygdom. Kan endvidere anvendes som rescue behandling – se Hypofysært CS.
ACTH-uafhængigt CS:
Medicinsk behandling med steroidgenese inhibitorer kan anvendes såfremt kirurgi ikke er mulig.
ACC: Der kan i tillæg til cytotoksisk kemoterapi, mitotane og adjuverende stråleterapi være indikation for andre steroidgenese inhibitorer mhp. at opnå eukortisolisme
Bilateral makronodulær hyperplasi: Såvel medicinsk som kirurgisk behandling kan komme på tale.
Behandling af komorbiditet
CS er associeret med en lang række komorbiditeter (inkl. kardiovaskulær sygdom, osteoporose, diabetes, psykiske symptomer), der bør behandles før, sideløbende og efter behandling af primær sygdom.
Der anbefales opmærksomhed på evt. indikation for perioperativ profylakse for venøs tromboemboli.
Ligeledes anbefales individuel vurdering mhp. indikation for influenza, herpes zoster og pneumokok vaccination.
Behandling af binyrebark-/hypofyse-insufficiens
GC-substitution
Glukokortikoid-substitution:
Ved opnåelse af remission er substitution med glukokortikoid indiceret indtil hypothalamus-hypofyse-binyre -aksen atter er intakt (ca. 6–12 måneder efter resektion af en ACTH-producerende tumor og ca. 18 måneder efter unilateral adrenalektomi – alle dog med stor variation). Patienter bør informeres om hyppig forekomst af glukokortikoid-withdrawal symptomer trods substitution med fysiologiske doser hydrokortison, og at det er forventeligt at en del vil føle sig dårligere tilpas uger til månederefter opnået remission. Withdrawal symptomer omfatter kvalme, anoreksi, vægttab og mere uspecifikke symptomer så som træthed, udmattelse, influenzalignende myalgier og artralgier. Nogle udvikler depression, angst og panik. Symptomlindring opnås ved at øge substitutionsdosis, men det er omvendt vigtigt at reducere dosis igen, når det er muligt for at undgå iatrogent CS.
Mineralokortikoid substitution:
Indiceret efter bilateral adrenalektomi, samt ved adrenolytisk behandling (se NBV om Binyrebarkinsufficiens)
Hypofyseinsufficiens:
Se NBV om hypofyseinsufficiens
Opfølgning
Generelt
CS i remission efter kirurgi kræver som udgangspunkt livslang kontrol grundet recidivrisiko specielt efter operation for hypofysær CS. Undtaget fra dette er oplagt benigne binyretumores (HU tomscanning på CT ≤ 10 og/eller oplagt benign histologi). Antallet af kontroller individualiseres men kan som udgangspunkt foregå årligt (UFC eller ONDST) – før ved klinisk mistanke om recidiv. Patienter i hydrokortisonsubstitution vurderes løbende klinisk og biokemisk for fortsat substitutions-behov ved synacthentest (evt. kun basal p-kortisol ved værdier < 138 nmol/l).
Endvidere fortsat kontrol og behandling af komorbiditeter opstået i forbindelse med CS.
Hypofysært CS
Livslang klinisk og biokemisk kontrol. Patienter, som postoperativt udvikler hypofyseinsufficiens, skal følges som andre patienter med denne tilstand. Det skal bemærkes, at patienter i hydrokortisonsubstitution også har en teoretisk risiko for udvikling af sent recidiv.
Efter hypofyseoperation/strålebehandling: Indikation for årlig kontrol af hypofysefunktion. Ved medicinsk behandling: kontrol i hht. anvendte præparaters bivirkningsprofiler.
Efter bilateral adrenalektomi: indikation for livslang årlig kontrol grundet risiko for Nelsons syndrom. Kontrollen vil typisk omfatte ACTH måling, vurdering af syn, og regelmæssig MR mhp. tumorstørrelse.
Ektopisk CS
Livslang klinisk og biokemisk kontrol.
Ved bilateral adrenalektomi foretaget ved mistanke om okkult sygdom må indikation for MR og ACTH kontrol overvejes, idet nogle patienter siden har vist sig at have hypofysært CS og ikke ektopisk sygdom.
ACTH-uafhængigt CS
Ved unilateralt oplagt benignt adenom (se ovenfor) er der ikke behov for recidivkontrol. Forløb opretholdes så længe der er behov for substitutionsbehandling. Ved ACC livslang kontrol og evt. adjuverende kemoterapi.
Visitation
CS er – udover den initiale udredning – tilknyttet en afdeling med højt specialiseret funktion.
Referencer
1. Lindholm J, Juul S, Jorgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, et al. Incidence and late prognosis of cushing’s syndrome: a population-based study. The Journal of clinical endocrinology and metabolism. 2001;86(1):117-23.
2. Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet (London, England). 2015;386(9996):913-27.
3. Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. The Journal of clinical endocrinology and metabolism. 2008;93(5):1526-40.
4. Newell-Price J, Trainer P, Perry L, Wass J, Grossman A, Besser M. A single sleeping midnight cortisol has 100% sensitivity for the diagnosis of Cushing’s syndrome. Clinical endocrinology. 1995;43(5):545-50.
5. John M, Lila AR, Bandgar T, Menon PS, Shah NS. Diagnostic efficacy of midnight cortisol and midnight ACTH in the diagnosis and localisation of Cushing’s syndrome. Pituitary. 2010;13(1):48-53.
6. Papanicolaou DA, Yanovski JA, Cutler GB, Jr., Chrousos GP, Nieman LK. A single midnight serum cortisol measurement distinguishes Cushing’s syndrome from pseudo-Cushing states. The Journal of clinical endocrinology and metabolism. 1998;83(4):1163-7.
7. Alwani RA, Schmit Jongbloed LW, de Jong FH, van der Lely AJ, de Herder WW, Feelders RA. Differentiating between Cushing’s disease and pseudo-Cushing’s syndrome: comparison of four tests. European journal of endocrinology. 2014;170(4):477-86.
8. Klose M, Kofoed-Enevoldsen A, Ostergaard Kristensen L. Single determination of plasma ACTH using an immunoradiometric assay with high detectability differentiates between ACTH-dependent and -independent Cushing’s syndrome. Scandinavian journal of clinical and laboratory investigation. 2002;62(1):33-7.
9. Ueland GA, Methlie P, Jossang DE, Sagen JV, Viste K, Thordarson HB, et al. Adrenal venous sampling for assessment of autonomous cortisol secretion. The Journal of clinical endocrinology and metabolism. 2018.
10. Young WF, Jr., du Plessis H, Thompson GB, Grant CS, Farley DR, Richards ML, et al. The clinical conundrum of corticotropin-independent autonomous cortisol secretion in patients with bilateral adrenal masses. World journal of surgery. 2008;32(5):856-62.
Tovholder
Claus Larsen Feltoft
Oprettet: Oktober 2018
Seneste revision: Oktober 2021
Næste revision: Oktober 2024
Øvrig arbejdsgruppe:
Ebbe Eldrup
Jakob Dal
Jakob Appel Østergaard
Jens Otto Lunde Jørgensen
Maciej Robaczyk
Marianne Andersen
Marianne Klose
Pernille Holmager
Åse Krogh Rasmussen
Interessekonflikter:
Ingen