Diagnosekode (ICD)
DE220
Definition
Akromegali: Klinisk tilstand fremkaldt af væksthormon (GH) overproduktion 1.
Gigantisme: Debut af akromegali før epifyseskiverne lukker 1.
Forekomst
Den årlige incidens er ca. 4 cases/106 indbyggere og prævalensen ca. 85 cases/106 indbyggere 1,2.
Ætiologi
Generelt:
Et hypofyseadenom med GH oversekretion er den hyppigste årsag. Ektopisk GHRH-secernerende tumor ses i sjældne tilfælde 1.
Arvelige former:
Akromegali er sjældent arveligt, men kan forekomme som led i multipel endokrin neoplasi (MEN) type 1 og 4 samt Carney complex (se boks 1+ MEN NBV). Mutation i aryl hydrocarbon receptor interacting protein (AIP)-genet kan medføre tidlig sygdomsdebut med en mere aggressiv variant med store hypofyseadenomer 3,4.
Symptomer og kliniske fund
Symptomer og fund kan relateres til tumortryk samt hormonel hypersekretion af GH eventuelt ledsaget af prolaktin (Ca. 30%, Hyperprolaktinæmi NBV) 1,5.
| Tumortryk | Synfeltsdefekter, hovedpine, trigeminus neuralgi, øjenmuskelparese/ptose, hypofyseinsufficiens |
| Fænotype | Hyperhidrose, fortykket hud, forgrovede ansigtstræk, prognatisme, diastema, tykke fingre, væskeretention (svampet håndtryk), brede fødder |
| Bevægeapparatet | Karpaltunnelsyndrom, polyartrose, artralgier, myalgier, vertebrale frakturer, neuropati, paræstesi |
| Kardiovaskulære manifestationer | Hypertension, hjerteinsufficiens, arytmier, kardiomyopati, aorta-og mitralpklapinssuficiens |
| Metaboliske effekter | Insulinresistens, nedsat glukose tolerance, diabetes mellitus, væskeretention |
| Respiratoriske komplikationer | Makroglossi, obstruktion af øvre luftveje/snorken, søvnapnø |
Som andre hypofyseadenomer kan også akromegali debutere klinisk som en ’pituitær apopleksi’ (akut indsættende hovedpine, nakke-rygstivhed, sløret sensorium, synsfeltsudfald, øjenmuskelparese, hypofyseinsufficiens)
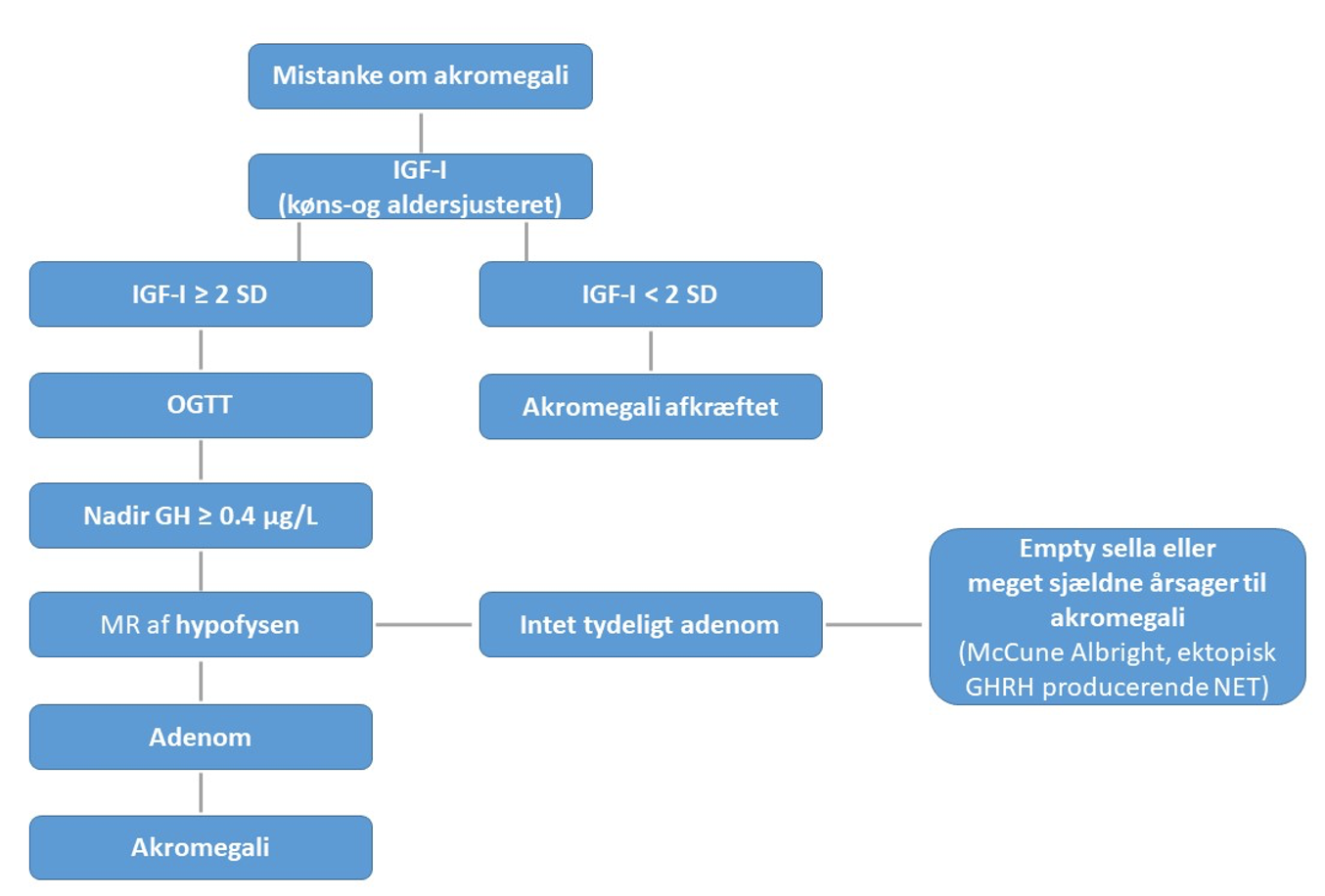
Udredning6 (Figur 1)
Indikation for udredning:
- Klassiske symptomer og akromegale træk
- Påvist hypofysetumor
Biokemisk udredning:
Køns- og alderskorrigeret serum-IGF-I anbefales som screeningstest. IGF-I inden for det normale referenceinterval vil med enkelte undtagelser udelukke diagnosen akromegali. Serum IGF-I koncentrationen er meget høj under og umiddelbart efter puberteten hos normalpersoner. Østrogenbehandling reducerer serum IGF-I koncentrationen, som også er lav i forbindelse med underernæring, leversygdom og visse andre kroniske sygdomme. Såfremt mistanken om akromegali opretholdes, henvises patienten til videre udredning på specialiseret enhed.
Den biokemiske diagnose konfirmeres ved manglende hæmning af GH sekretion ved en 75 g oral glukose toleranstest (OGTT). OGTT kan anvendes ved velreguleret diabetes mellitus. Nadir GH efter OGTT < 0,4 µg/L og normal IGF-I udelukker diagnosen. Serielle GH målinger med eksempelvis 30. min interval over 2 timer kan anvendes som alternativ hvor nadir GH < 1 µg/L vil betragtes som normale.
Billeddiagnostisk udredning:
Ved klinisk og biokemisk påvist akromegali suppleres med billeddiagnostisk udredning. MR skanning af hypofysen med kontrast anbefales, alternativt CT-skanning, hvis MR er kontraindiceret. Såfremt der ikke påvises hypofysetumor, kan ekstrapituitær akromegali overvejes. Udredning vil ofte inkludere måling af plasma GHRH suppleret af billeddiagnostik.
Undersøgelser ved påvist hypofysetumor:
- Neuro-oftalmologisk vurdering ved mistanke om affektion af chiasma opticum
- Hypofysefunktion: Prolaktin cosekretion (evt. TSH cosekretion) og hypofyseinsufficiens
Øvrige undersøgelser:
- Udredning for komorbiditet (BT, EKG, ekkokardiografi, HbA1c, screening for søvnapnø, DXA-skanning)
- Genetisk screening for AIP mutation anbefales ved positiv familieanamnese og bør overvejes ved tidlig sygdomsdebut (før 30-årsalderen) med tilstedeværelse af et stort hypofyseadenom 4,7, eventuel screening for MEN1 og MEN44.
Figur 1: Udredningsalgoritme ved klinisk mistanke om akromegali. *enkelte undtagelser betinget af medicinsk behandling eller kronisk sygdom
Primære behandlingsmål6,8
Formål med behandlingen:
1) Biokemisk sygdomskontrol med normalisering af GH og IGF-I koncentrationerne
2) Kontrol af tumorvækst
3) Substitution af hypofyseinsufficiens
Biokemisk sygdomskontrol:
Spot GH koncentration < 1 μg/L eller nadir GH < 0,4 μg/L under en glukosebelastning og normaliseret IGF-1 koncentration i forhold til aldersrelateret normalområde.
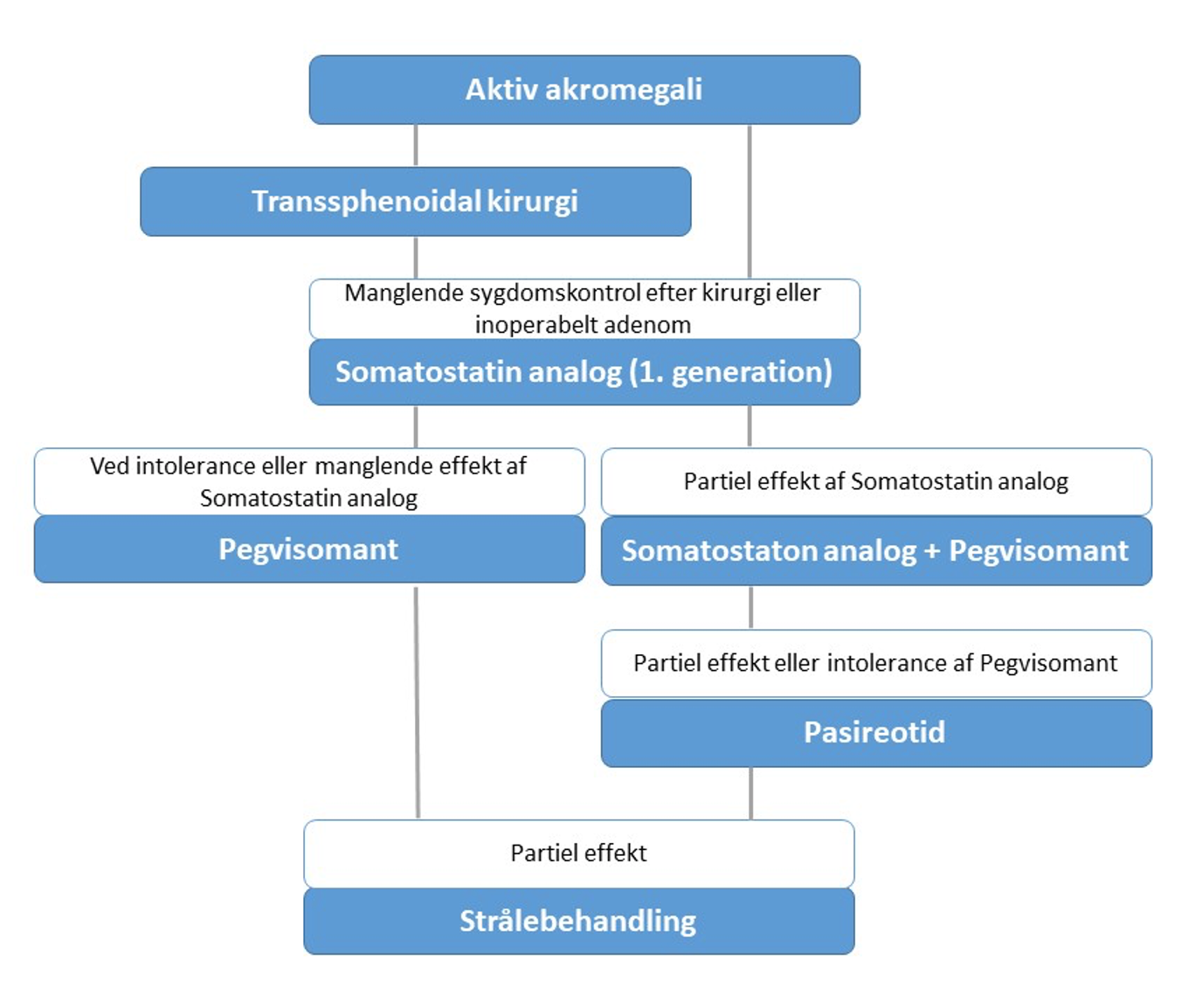
Behandling (Figur 2)
Behandling af akromegali varetages på højt specialiseret endokrinologisk afdeling 6,9,10.
- Kirurgi i form af transsfenoidal adenomektomi er førstevalg
- Medicinsk behandling: Anvendes primært postoperativt ved persisterende sygdomsaktivitet. Kan anvendes som primær behandling hos patienter, hvor kirurgi ikke er mulig, skønnes virkningsløs eller ved patientønske.
Somatostatin analoger:
Somatostatin-analogerne Lanreotid (Ipstyl Autogel®) og Octreotid (Sandostatin Lar®) er første-linje medicinsk behandling. Ipstyl startdosis er 60 – 90 mg dybt s.c. hver 4. uge, ens startdosis forSandostatin Lar® er 10 – 20 mg i.m. hver 4 uge. Doserne kan i udvalgte tilfælde øges ud over de vanlige maksimale doser. Pasireotid (Signifor®) er en nyere og mere potent somatostatin analog, som især hæmmer somatostatinreceptor subtype 5. Det er mere effektivt end Lanreotid og Octreotid med hensyn til at reducere serum IGF-I, men også forbundet med en væsentlig større risiko for behandlingskrævende diabetes mellitus grundet en til tider udtalt hæmning af insulin sekretionen. Pasireotid overvejs ved manglende effekt af Pegvisomant (se nedenfor). Det er særdeles vigtigt at monitorere den glykæmiske status især de første 1-6 måneder, idet ketoacidose kan opstå.
Dopamin agonister
Cabergolin® er effektivt hos relativt få patienter, og er sjældent sufficient som monoterapi. Startdosis er 0,5 mg Cabergolin 2 gange om ugen.
Væksthormonreceptorantagonist:
Pegvisomant (Somavert®) er en specifik GH antagonist . Behandlingen er effektiv til at reducere IGF-I. I Danmark anvendes Pegvisomant primært i kombinationsbehandling med SA, hvor der ikke ses tilstrækkelig klinisk eller biokemisk respons på SA monoterapi. Ved kombinationsbehandling optræder stigning i koncentrationen af levertransaminase hos 10-15 % af patienterne. Derfor måles ALAT regelmæssigt initialt med 1-3 måneders interval og herefter hver 6. måned. Pegvisomant kan tillige anvendes som monoterapi i tilfælde hvor kirurgisk kontrol ikke kan opnås og hvor behandling med SA er enten helt virkningsløs eller ikke tåles. Startdosis er 15 mg 2 gange ugentligt ved kombinationsbehandling med en somatostatin analog, hvor en pevosomantdosis på 40-60 mg pr. uge fordelt på 2 injektioner ofte er tilstrækkelig. Bemærk at pegvisomantbehandling medfører en stigning i serum GH niveauet, hvorfor den biokemiske kontrol alene baseres på serum IGF-I niveauet.
- Strålebehandling: Generelt kun indiceret i sjældne tilfælde med manglende sygdomskontrol efter kirurgisk og medicinsk behandling.
Figur 2: Simplificeret behandlingsalgoritme ved aktiv akromegali. Behandlingsmodaliteter som præoperativ forbehandling med en somatostatin analog for at opnå skrumpning samt behandling med dopamin agonist er ikke anført, men kan være relevante behandlinger i udvalgte tilfælde.
Opfølgning
De fleste patienter følges livslangt i et endokrinologisk specialambulatorium jf. Sundhedsstyrelsens retningslinjer.
Klinisk og biokemisk opfølgning 6
Efter hypofysekirurgi:
undersøges patienten for diabetes insipidus og anterior hypofyseinsufficiens. Indikation for hydrokortisonbehandling umiddelbart postoperativt afhænger af symptomer og lokal praksis.
Patienten følges med ACTH-test og hypofysestatus ca. 6 uger postoperativt.Vurdering af væksthormonaksen foretages igen ca.3 måneder efter operationen.
For alle behandlingsmodaliteter gælder, at der ved tilfredsstillende klinisk og biokemisk respons foretages klinisk undersøgelse, biokemisk kontrol af sygdomsaktivitet og undersøgelse af øvrige hypofyseakser med passende interval. Intervaller af 1 – 2 års længde vil være typisk. Strålebehandling medfører stor risiko for udvikling af hypofyseinsufficiens, der kan optræde mange år efter endt behandling.
Tumoropfølgning
MR-skanning med kontrast og oftalmologisk undersøgelse udføres på individuel basis ved mistanke om tumorvækst.
Behandling og kontrol af co-morbiditet 10-13
Glukose metabolisme: HbA1c
Cancersygdomme: Akromegali er associeret med en øget cancerrisiko, hvorfor patienter med akromegali bør opfordres til at følge de nationale screeningsprogrammer for colon- og mammacancer, og særlig opmærksomhed bør henledes på cancersuspekte symptomer.
Hjertesygdomme: BT og lipider måles ved de kliniske kontroller, og forhøjede værdier behandles efter vanlige kriterier. EKG, evt. ekkokardiografi, udføres ved mistanke om kardiomegali og ve. ventrikeldysfunktion.
Søvnforstyrrelser: Undersøgelse for søvnapnø i tilfælde af relevante symptomer (snorken, dagstidssomnolens).
Osteoporose: DXA-skanning anbefales før behandlingsstart og kan overvejes når der er opnået acceptabel sygdomskontrol. Man skal være opmærksom på, at der er en øget risiko for vertebrale frakturer hos patienter med akromegali, også selvom der ikke er påvist osteoporose ved DXA-skanning. Udredning og behandling for osteoporose følger standard retningslinjer (Postmenopausal osteoporose NBV & Mandlig osteoporose NBV).
Prognose ved akromegali
Aktiv akromegali er forbundet med øget morbiditet og mortalitet navnlig relateret til den øgede forekomst af hjerte- karsygdom. Ved opnåelse af biokemisk kontrol (se ovenfor) er det tidligere vist, at mortaliteten kan reduceres svarende til baggrundsbefolkningen. Biokemisk kontrol efter kirurgi opnås hos 50 – 60% og er meget afhængig af tumorstørrelse på operationstidspunktet. Behandling med somatostatin analog er effektiv hos cirka 60 %, og hos cirka halvdelen opnås tillige signifikant reduktion af tumorstørrelse. Ved Pegvisomant-behandling ses normalisering af IGF-I hos 75 – 95 % efter 18 – 24 måneder, men ingen reduktion af tumorstørrelsen2,14-16.
Referencer
1. Ben-Shlomo A, Melmed S. Acromegaly. Endocrinol Metab Clin North Am. 2008;37(1):101-122, viii.
2. Dal J, Feldt-Rasmussen U, Andersen M, et al. Acromegaly incidence, prevalence, complications and long-term prognosis: a nationwide cohort study. Eur J Endocrinol. 2016;175(3):181-190.
3. Boguslawska A, Korbonits M. Genetics of Acromegaly and Gigantism. J Clin Med. 2021;10(7).
4. Lecoq AL, Kamenicky P, Guiochon-Mantel A, Chanson P. Genetic mutations in sporadic pituitary adenomas–what to screen for? Nat Rev Endocrinol. 2015;11(1):43-54.
5. Colao A, Ferone D, Marzullo P, Lombardi G. Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev. 2004;25(1):102-152.
6. Katznelson L, Laws ER, Jr., Melmed S, et al. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(11):3933-3951.
7. Cazabat L, Bouligand J, Salenave S, et al. Germline AIP mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab. 2012;97(4):E663-670.
8. Giustina A, Chanson P, Bronstein MD, et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab. 2010;95(7):3141-3148.
9. Giustina A, Chanson P, Kleinberg D, et al. Expert consensus document: A consensus on the medical treatment of acromegaly. Nat Rev Endocrinol. 2014;10(4):243-248.
10. Melmed S, Bronstein MD, Chanson P, et al. A Consensus Statement on acromegaly therapeutic outcomes. Nat Rev Endocrinol. 2018;14(9):552-561.
11. Melmed S, Casanueva FF, Klibanski A, et al. A consensus on the diagnosis and treatment of acromegaly complications. Pituitary. 2013;16(3):294-302.
12. Jenkins PJ. Cancers associated with acromegaly. Neuroendocrinology. 2006;83(3-4):218-223.
13. Dal J, Leisner MZ, Hermansen K, et al. Cancer Incidence in Patients With Acromegaly: A Cohort Study and Meta-Analysis of the Literature. J Clin Endocrinol Metab. 2018;103(6):2182-2188.
14. Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical ‘cure’. Eur J Endocrinol. 2005;152(3):379-387.
15. Dekkers OM, Biermasz NR, Pereira AM, Romijn JA, Vandenbroucke JP. Mortality in acromegaly: a metaanalysis. J Clin Endocrinol Metab. 2008;93(1):61-67.
16. Holdaway IM, Bolland MJ, Gamble GD. A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol. 2008;159(2):89-95.
Boks 1 – genetiske årsager til akromegali
| Gen | Arvegang | Karakteristisk | |
| Multipel endokrin neoplasi type 1 | MENIN | Aut.dom | Primær hyperparathyroidisme, entero-pankreatiske tumorer, hypofyseadenomer m.fl. (se MEN-NBV) |
| Multipel endokrin neoplasi type 4 | CDKN1B | Aut.dom | Primær hyperparathyroidisme og hypofyseadenomer (og andre tumorer) |
| Carney’s complex syndrom | PRKAR1A | Aut.dom | Pigmentering (plettet), myxom, testikel-, binyre- og hypofyseadenomer eller hyperplasi |
| McCune-Albright syndrome | GNAS1 | Nedarves ikke – sporadisk mutation | Fibrøs dysplasi, cafe-au-lait pletter, endokrinopati |
| Familiære isolerede hypofyseadenomer | AIP (udgør 20% af tilfældene) | Aut.dom | Hypofyseadenomer (hyppigst med somatotropinomer eller prolaktinomer) |
| X-linked Acrogigantism (X-LAG) | Duplikation af kromoson Xq26.3, GPR101 | X-bunden dominant | Hypofyseadenom,debut før 2 års alderen (oftest GH- eller PRL-adenom) |
Tovholdere
Mai Christiansen Arlien-Søborg og Jens Otto L. Jørgensen
Øvrige forfattere:
Marianne Andersen
Jakob Dal
Marianne Klose
Mikkel Andreassen
Eigil Husted Nielsen
Caroline Nervil Kistorp
Ulla Feldt-Rasmussen
Christian Rosendal
Revideret: oktober 2021
Næste revision: oktober 2024