Hvad omfatter denne NBV?
Diagnostik, behandling og opfølgning af patienter med Maturity-Onset Diabetes of the Young (MODY)
Diagnostik, behandling og opfølgning af andre patienter med diabetes som skyldes sygdomsfremkaldende varianter i gener der er involveret i MODY.
MODY og graviditet
Diagnostik, behandling og opfølgning af patienter med Neonatal Diabetes (NDM) herunder Transient (TNDM) og Permanent Neonatal Diabetes (PNDM)
Diagnostik, behandling og opfølgning af patienter med Maternally Inherited Diabetes and Deafness (MIDD)
Hvad omfatter denne NBV ikke?
Monogenetiske insulin resistens syndromer; monogenetiske lipodystrofier; syndromiske diabetesformer med andre genetiske ætiologier end HNF4A/1A/1B, ABCC8 og KCNJ11 samt 6q24 abnormiteter; opstart og justering af insulinbehandling hos spædbørn. Denne NBV omfatter ikke genetiske ætiologier specifikke for Grønland.
Diagnosekoder (ICD)
Andre former for diabetes DE13
Neonatal diabetes mellitus P70.2
Ved MIDD evt.:
Mitokondriel myopati DG71.3
Forkortelser
AD Autosomal dominant
ADHD Attention deficit hyperactivity disorder
AR Autosomal recessive
CK Creatinkinase
CPEO Kronisk progressiv ekstern oftalmoplegi
DEND Developmental delay, epilepsy and neonatal diabetes
DKA Diabetisk ketoacidose
DPP-IV Dipeptidylpeptidase–IV hæmmer
F-BG Faste blodglukose
GDM Gestationel diabetes mellitus
GLP1 Glucagon-like peptide-1
GSIS Glukose stimuleret insulin sekretion
iDEND Intermediate DEND
IS Insulinfølsomhed
IUGR Intrauterine growth retardation
MDI Multiple daglige injektioner
MELAS Myopati, encefalopati, laktat-acidose og stroke-like episoder
MIDD Maternally inherited diabetes and deafness
MODY Maturity-onset diabetes of the young
NDM Neonatal diabetes mellitus
OGTT Oral glukose tolerance test
PNDM Permanent neonatal diabetes mellitus
SGLT-2i Sodium-glucose co-transporter-2 inhibitor
SU SulfonylurinstofTNDM Transient neonatal diabetes mellitus
Definition
Monogenetisk diabetes er en samlebetegnelse for diabetesformer, som ofte skyldes en eller flere defekter i et gen, som er involveret i udvikling og/eller funktionen af de pankreatiske betaceller. Monogenetisk diabetes kan ses i familier med dominant, recessiv eller non-mendelsk arvegang eller opstår spontant som følge af en de novo mutation. MODY, MIDD og NDM repræsenterer 3 forskellige typer af monogenetisk diabetes. Øvrige monogenetiske diabetesformer inkluderer syndromer, hvor diabetes er en af flere organmanifestationer.
Genetisk diagnosticering er vigtig fordi behandlingen af forskellige subtyper af monogenetisk diabetes kan afvige fra behandling af type 1-diabetes og type 2-diabetes. Endvidere muliggør en genetisk diagnose genetisk rådgivning, herunder familieudredning, samt kvalificeret prognosticering af fremtidig risiko for sen-diabetiske komplikationer og behandlingsbehov for den enkelte patient.
Tabel 1:Differentiering mellem type 1-, type 2-diabetes og MODY
| MODY | Type 1-diabetes | Type 2- diabetes | |
| Antal gener involveret for den enkelte patient | 1 gen (monogen) | Polygen, HLA-genet og andre | Polygen, multiple gener |
| Arv | 50 % videregiver mutation til børn (autosomal dominant arvegang) | Forøget risiko hvis forældre har type 1-diabetes. Miljøfaktorer spiller ind | Forøget risiko hvis forældre har type 2-diabetes. Miljøfaktorer spiller ind |
| Patofysiologi | Påvirket betacelle funktion/udvikling | Autoimmun destruktion af betaceller | Insulinresistens, relativ insulinmangel |
| Alder ved debut | Ofte <25 år (patient eller diabetiske slægtninge) | Typisk 8-12 år, men principielt hele livet | Typisk > 40 år |
| Kropsvægt | Som baggrundsbefolkningen | Som baggrundsbefolkningen | Normal-overvægtig |
| Autoantistoffer | Som rask del af baggrundsbefolkningen | 90 % ved debut | Som rask del af baggrundsbefolkningen |
| C-peptid 3-5 år efter diabetesdiagnose | Målbar, men typisk lavt i normalområdet eller under | Umålelig-meget lav | Normal-høj |
| Association med det metaboliske syndrom | Nej | Nej | Ja |
| Association med anden autoimmun sygdom | Nej | Ja | Nej |
MODY
Forekomst
Maturity-Onset Diabetes of the Young (MODY) udgør 1-3 % af diabetes patienter, hvor mere end 50 % af patienterne på nuværende tidspunkt er (mis)-klassificerede som havende enten type 1- eller type 2-diabetes (1).
Ætiologi
Den genetiske ætiologi til MODY er heterogen, og til dato er beskrevet 14 forskellige gener, hvori defekter er forbundet med en MODY fænotype (tabel 1). Autosomal dominante mutationer i HNF4A (MODY1), GCK (MODY2), HNF1A (MODY3) samt HNF1B (MODY5) udgør tilsammen langt de hyppigste genetiske ætiologier, og penetransen for patogene varianter er op mod 90 %. De øvrige 10 MODY subtyper er sjældne. Patienter med transient neonatal diabetes kan udvikle diabetes igen omkring pubertetens indtræden, hvor andre gener er involverede (se særskilt afsnit om neonatal diabetes (NDM)).
Tabel 1. Oversigt over MODY typer, gen og hyppighed.
| MODY type | Prævalens blandt patienter med MODY (%) | Original reference |
| HNF4A-MODY (MODY1) | 5 | https://www.ncbi.nlm.nih.gov/pubmed/8945471 |
| GCK-MODY (MODY2) | 30-50 | https://www.ncbi.nlm.nih.gov/pubmed/1545870 |
| HNF1A-MODY (MODY3) | 30-50 | https://www.ncbi.nlm.nih.gov/pubmed/8945470 |
| IPF1-MODY (MODY4) | <1 | https://www.ncbi.nlm.nih.gov/pubmed/9326926 |
| HNF1B-MODY (MODY5) | 5 | https://www.ncbi.nlm.nih.gov/pubmed/9398836 |
| NEUROD1-MODY (MODY6) | <1 | https://www.ncbi.nlm.nih.gov/pubmed/10545951 |
| KLF11-MODY (MODY7)* | <1 | https://www.ncbi.nlm.nih.gov/pubmed/15774581 |
| CEL-MODY (MODY8) | <1 | https://www.ncbi.nlm.nih.gov/pubmed/16369531 |
| PAX4-MODY (MODY9)* | <1 | https://www.ncbi.nlm.nih.gov/pubmed/17426099 |
| INS-MODY (MODY10) | <1 | https://www.ncbi.nlm.nih.gov/pubmed/18162506 |
| BLK-MODY (MODY11)* | <1 | https://www.ncbi.nlm.nih.gov/pubmed/19667185 |
| ABCC8-MODY (MODY12) | <1 | https://www.ncbi.nlm.nih.gov/pubmed/21989597 |
| KCNJ11-MODY (MODY13) | <1 | https://www.ncbi.nlm.nih.gov/pubmed/22701567 |
| APPL1-MODY (MODY14) | <1 | https://www.ncbi.nlm.nih.gov/pubmed/26073777 |
*Nyligt studie peger på at KLF11, PAX4 og BLK ikke er MODY-gener og de forventes at udgå fra genetisk udredning for MODY (Laver TW et al., 2021, preprint).
Symptomer og kliniske fund
Ved følgende kliniske karakteristika hos en diabetespatient bør en genetisk udredning for MODY overvejes (se figur 1):
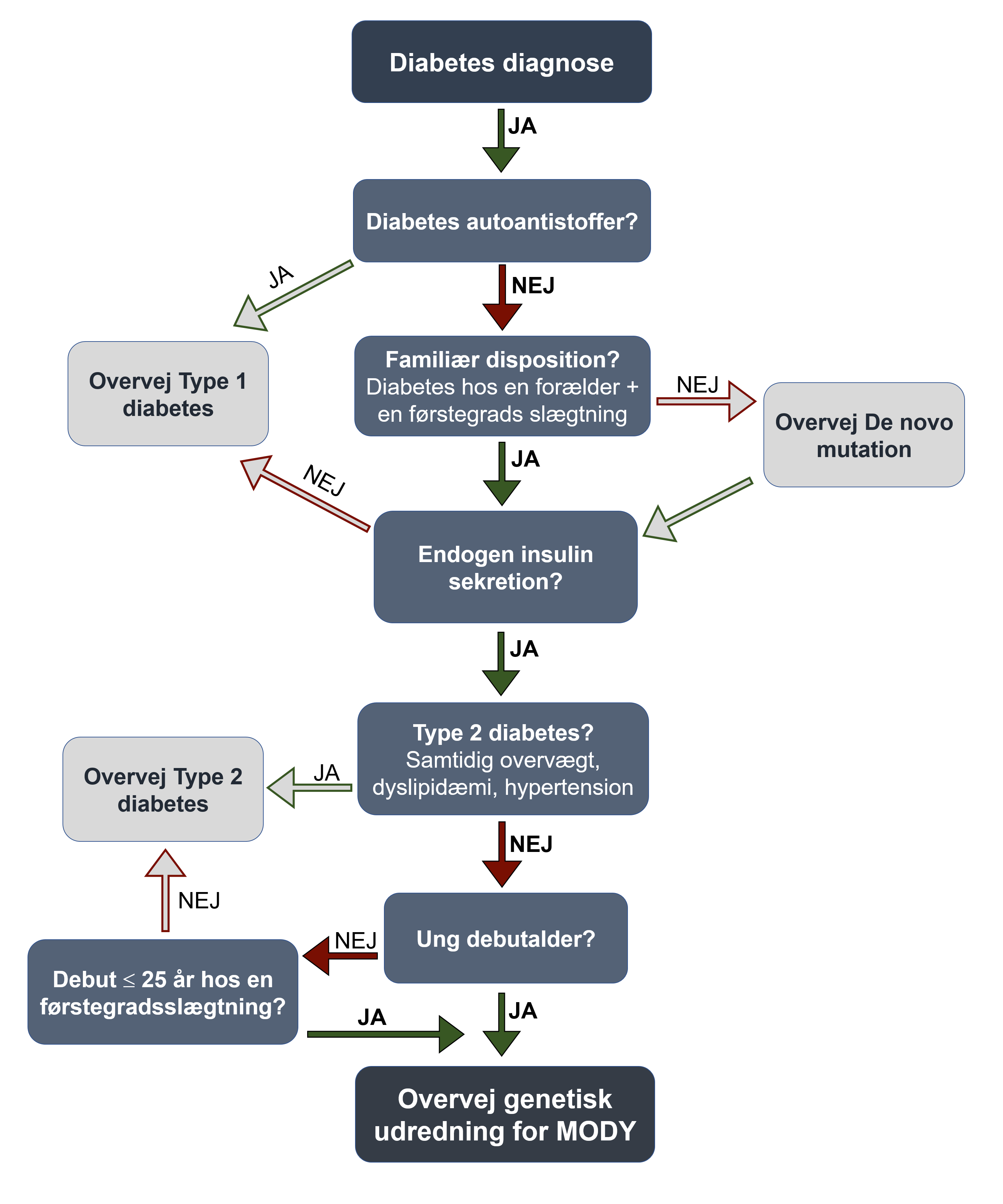{kind=link}
- Hvis en forældre og/eller en anden førstegradsslægtning har diabetes
- Fravær af type 1-diabetes karakteristika (dvs. fravær af pankreatiske autoantistoffer og bevaret endogen insulin produktion 3-5 år efter diagnosen (målbart C-peptid/insulin-uafhængig/beskedent insulin-behov))
- Fravær af type 2-diabetes karakteristika (overvægt, andre komponenter af det metaboliske syndrom)
- diabetes-debut før 25-års alderen i patienten og/eller diabetiske slægtninge
Hos patienter som alene opfylder punkterne 2-4 bør muligheden for en de novo mutation haves i mente og genetisk udredning bør overvejes.
MODY kan præsentere sig på flere måder:
- Tilfældigt fund af hyperglykæmi i tilslutning til graviditet, helbredsundersøgelser, screeningsundersøgelser
- Symptomgivende hyperglykæmi (vægttab, tørst, øget vandladning, træthed, synsforstyrrelser, lægkramper, føleforstyrrelser, kløe, genitale svampeinfektioner)
Tabel 2: Oversigt over kliniske fund ved de 4 hyppigste MODY subtyper
| Type | Patofysiologi | Fænotype | Biokemi | Øvrige fænotype | Behandling |
| HNF4A-MODY (MODY1) | Betacelle dysfunktion med normal IS, påvirket GSIS pga. mangelfuld betacelle depolarisering | Progredierende, symptomgivende hyperglykæmi, normal kropsvægt Risiko for komplikationer, tidlig udvikling af diabetisk retinopati | F-BG: let forhøjet-høj HbA1c: sjældent>100 mmol/mol OGTT: 120-min. glukosestigning>5 mmol/l
| Neonatal hyperinsulinaemisk hypoglykæmi (~15%), makrosomi (fødselsvægt > 4 kg ses hos ~50 %). Fanconi syndrom (hypercalciuri og nefrocalcinose) Se afsnit om graviditet | Diæt SU Insulin |
| GCK-MODY (MODY2) | Højre-forskudt dosisresponskurve for GSIS, normal IS
| Mild, ikke-progredierende hyperglykæmi til stede fra fødslen, asymptomatisk, normal kropsvægt Let øget forekomst af simplex retinopati, ingen risiko for øvrige komplikationer | F-BG: 5,4-8,3 mmol/l HbA1c: 38-60 mmol/mol OGTT: 120-min. glukosestigning<3 mmol/l
| Se afsnit om graviditet | Intet behandlingsbehov udenfor graviditet |
| HNF1A-MODY (MODY3) | Betacelle dysfunktion, normal IS, påvirket GSIS pga. mangelfuld betacelle depolarisering | Progredierende, symptomgivende hyperglykæmi, normal kropsvægt Risiko for komplikationer, tidlig udvikling af diabetisk retinopati | F-BG: normal-høj HbA1c: sjældent>100 mmol/mol OGTT: 120-min glukosestigning>5 mmol/l Andet: HDL-C ofte>1,2 mmol/l, glukosuri
| Benigne hepatocellulære adenomer
| Diæt SU Repaglinid til måltiderne DPP-IV hæmmer GLP1-analog Insulin |
| HNF1B-MODY (MODY5) | Normal IS, nedsat insulin sekretion | Symptomgivende hyperglykæmi, DKA kan forekomme, 30 % de novo mutationer Risiko for komplikationer. 50 % udvikler dialyse-krævende nefropati før 45-års alderen | F-BG: variabel HbA1c: variabel OGTT: — Andet: i nogle tilfælde påvirket leverbiokemi, forhøjet s-urat, hypomagnesiæmi | Varierende sværhedsgrad af ekstra-pankreatiske manifestationer i nyre-urinveje og genitalia, pankreas atrofi (se tekst)
| Insulin |
Udredning
Ved klinisk mistanke om MODY suppleres med bestemmelse af et ikke-fastende C-peptid med matchende BG, GAD65 antistof og eventuelt ø-celle antistoffer (hvis ikke tidligere bestemt). Hvis patienten har bevaret betacellefunktion og negative diabetes autoantistoffer, tilbydes genetisk udredning (figur 1). Til støtte i den kliniske beslutningsproces kan eventuelt anvendes en MODY sandsynlighedsberegner (https://www.diabetesgenes.org/mody-probability-calculator/).
Som udgangspunkt bør kun patienter med tilknytning til et diabetesambulatorium, tilbydes genetisk udredning. Den genetiske udredning af patienter med MODY varetages forskelligt i landets 5 regioner:
Region Hovedstaden, Sjælland og Nord: Steno Diabetes Center Copenhagen (SDCC) https://www.sdcc.dk/for-fagfolk/salg-af-analyser/Sider/MODY-diagnostik.aspx
Region Syd: Klinisk Genetisk Afdeling, Odense Universitetshospital (http://www.ouh.dk/wm504172)
Region Midt: SDCC eller Molekylær Medicinsk Afdeling, AUH (https://www.auh.dk/afdelinger/molekylar-medicinsk-afdeling/)
Relevant biologisk materiale, typisk en EDTA fuldblodsprøve, sendes sammen med kliniske oplysninger til den diagnostiske afdeling. En positiv diagnostisk test bør følges op af en familieudredning af førstegradsslægtninge, hvor familiemedlemmer undersøges for, om de er bærere af familiens mutation. Øvrige familiemedlemmer opfordres til at opsøge genetisk udredning (i særdeleshed familiemedlemmer med diabetes) via praktiserende læge der kan henvise til lokalt diabetesambulatorium. Prisen for den genetiske udredning er faldet betydeligt de senere år og kan i flere genetiske laboratorier tilbydes for under 5000 kr. ved undersøgelse af de 4 hyppigste MODY subtyper.
Patienter som opfylder MODY-kriterier (beskrevet under ‘Symptomer og fund’) vil i fremtiden sandsynligvis kunne tilbydes test via det Nationale Genom Center (henvisningskriterier og arbejdsgange er aktuelt under udarbejdelse).
Prognose
Se tabel 2. Ligesom i anden diabetesbehandling er god glykæmisk kontrol og reduktion af øvrige kardiovaskulære risikofaktorer essentiel for en god prognose.
Behandling
- Kolesterolreducerende behandling: Ingen evidens foreligger vedrørende kolesterolreducerende behandling. Der anbefales en tilgang svarende til behandling af type 1-diabetes (link T1DM nbv). For patienter med GCK-MODY (MODY2) følges dog retningslinjer for den raske baggrunds befolkning.
- Blodtrykssænkende behandling: Ingen studier foreligger vedrørende blodtrykssænkende behandling, men behandlingsmål som ved type 1-diabetes anbefales. For patienter med GCK-MODY (MODY2) følges retningslinjer for patienter med ukompliceret essentiel hypertension.
- Glukosereducerende behandling: Ingen langtidsstudier viser evidens for effekten af glukosereducerende behandling, men der bør stiles mod glykæmisk kontrol svarende til anbefalinger for patienter med type 1-diabetes (link T1DM nbv). Patienter med GCK-MODY skal ikke have glukosereducerende behandling uden for graviditet (se nedenfor)(2). Patienter med MODY er ikke beskyttet mod udvikling af andre diabetesformer. Ved vedvarende behandlingssvigt bør der i tillæg til vurdering af behandlings-compliance overvejes undersøgelse for udvikling af anden diabetestype (3).
- HNF4A-MODY (MODY1)
- Førstevalg er sulfonylurinstoffer (SU), se HNF1A-MODY (MODY3) for nærmere beskrivelse
- Alternativer: Øvrige antidiabetiske stofklassers effekt er ikke testet i HNF4A-MODY (MODY1)
- GCK-MODY (MODY2)
- Skal som udgangspunkt ikke behandles uden for graviditet. Se under ”graviditet” for håndtering
- Ved HbA1c >60 mmol/mol hos patient over 40 år og >56 mmol/mol hos patient under 40 år, overvejes om patienten har udviklet anden diabetes i tillæg til GCK-MODY (MODY2) [6]
- HNF1A-MODY (MODY3)
- Peroral behandling er at foretrække frem for insulinbehandling. Selv ved langvarig insulinbehandling kan størstedelen af patienter overgå til SU med færre udsving i BG og optimeret HbA1c til følge. Dette er dokumenteret ved insulindoser mellem 0,5 til 2,2 enheder/kg kropsvægt i døgnet. Ved utilstrækkelig glukosereducerende effekt af SU som monoterapi, kan nogle patienter have et behov for supplerende behandling.
- Førstevalgsbehandling er langtidsvirkende SU (fx Glimepirid) (4, 5). Initialt 0,5 mg Glimepirid dagligt med dosisøgning med 0,5 mg ad gangen hver 2. uge. Ved udtalt hyperglykæmi evt. dosisøgning hver uge. Vedligeholdelsesdosis sædvanligvis 2-4 mg dagligt. Ved skifte fra insulinbehandling til SU-behandling, foreslås følgende tilgang: Patienter med total døgn dosis insulin < 20 enheder: insulin seponeres uden udtrapning og Glimepirid startes og justeres som beskrevet ovenfor. Patienter med total døgn dosis >20 enheder: insulindosis halveres hver 2. uge samtidigt med at Glimepirid dosis øges med 0,5 mg, således at behandlingsskiftet sker over uger. Patienterne informeres om risiko for hypoglykæmi specielt i tilslutning til faste og motion (4). Patienterne informeres ligeledes om insulinbehandling ved evt. fremtidige graviditeter og at insulin i nogle tilfælde kan blive nødvendig igen efter lang sygdomsvarighed eller ved tilstødende komorbiditet.
- Ved recidiverende hypoglykæmi mellem måltiderne, kan Repaglinid anvendes som alternativ til SU. Initialt Repaglinid 0,5 mg 15 minutter før hvert hovedmåltid. Dosisjustering hver 2. uge. Maksimal dosis 4 mg 3 gange dagligt (6)
- Metformin har en begrænset plads i behandlingen af yngre patienter, men kan eventuelt anvendes ved behandling af insulinresistente patienter (5)
- GLP1-agonister kan anvendes som alternativ til SU ved problemer med hypoglykæmi. Fx Liraglutid 0,6 mg s.c. 1 gang dagligt i mindst 1 uge og herefter dosisøgning til 1,2 mg s.c. dagligt og efter yderligere en uge dosisøgning til 1.8 mg s.c. dagligt (7)(8)
- DPP-IV-hæmmere kan anvendes sammen med SU eller som alternativ til SU ved problemer med hypoglykæmi. Ved kombinationsbehandling kan dosisreduktion af SU overvejes (9)
- Der er ingen klinisk erfaring med anvendelse af SGLT2i
- HNF1B-MODY (MODY5)
- Tidlig insulinbehandling, ofte MDI (10)
Behandling af MODY under graviditet
En del kvinder med MODY vil få stillet deres diabetesdiagnose i tilslutning til graviditet via screening for GDM og ved påvist diabetes (mis-)klassificeres de ofte som havende regelret GDM. I forhold til betydningen af diabetes hos den gravide samt diabetesbehandlingen i graviditeten med insulin og kostvejledning henvises til NBV ’Diabetes og Graviditet’.
I Danmark anbefales insulinbehandling til gravide med MODY (se dog særlige forhold vedr. GCK-MODY nedenfor). Patienter som tabletbehandles forud for graviditet, omstilles til insulinbehandling allerede når graviditet planlægges, og fortsættes indtil efter fødslen og evt. amning er ophørt.
GCK-MODY (MODY2)
Behandling af GCK-MODY (MODY2) kan være påkrævet under graviditeten og kan i modsætning til uden for graviditeten være forbundet med betydelige udfordringer. Påvirkningen af fostrets vækst afhænger af, om fosteret er bærer af en GCK mutation. Insulin er en central intrauterin vækstfaktor og fostervæksten kan påvirkes, hvis fosterets tærskelværdi for insulinsekretion er forskelligt fra moderens og/eller hvis moderens blodglukose (insulinbehandling) holdes for lavt til at stimulere insulinsekretion i fosteret (jf. tabel 3). Under en GCK-MODY (MODY2) graviditet er fosterets bærerstatus af mutationen ukendt og indtil videre anbefales tæt kontrol af fosterets vækst ved ultralydsscanning. Hvis fostrets abdominale omfang overstiger 75-percentilen, anbefales det, at insulinbehandling af moderen påbegyndes (11). Insulin-behandling ved GCK-MODY (MODY2) graviditeter har ikke dokumenteret effekt i forhold til at begrænse fosterets øgede vækst (i de tilfælde hvor fosteret ikke har arvet mutationen), men anbefales indtil videre alligevel. Igangsættelse af fødsel kan overvejes fra uge 38. Væksthæmning af et foster som har arvet sin mutation fra faderen kan være diagnostisk udfordrende såfremt tilstanden ikke er erkendt hos faderen og/eller obstetrikeren ikke er vidende om faderens bærer-tilstand.
Tabel 3. Fosterets vækst under MODY2 graviditet
| Foster +MODY2 | Foster –MODY2 | |
| Mor +MODY2 | Normal vækst uden behandling (risiko for væksthæmning ved behandling) | Risiko for øget vækst |
| MOR –MODY2 | Risiko for væksthæmning | – |
HNF4A-MODY (MODY1)
15% af patienter med HNF4A-MODY (MODY1) har et bifasisk forløb med behandlingskrævende hyperinsulinæmisk neonatal hypoglykæmi samt udvikling af diabetes senere i livet. Hypoglykæmien kan være udtalt og sammenhængen med HNF4A-MODY kan være forbundet med et diagnostisk delay, specielt såfremt mutationen nedarves fra faderen hvor den familiære disposition til diabetes kan blive overset af behandlerne.
Visitation og opfølgning
MODY-patienter bør følge diabeteskontroller i et diabetes ambulatorium svarende til, hvad der tilbydes patienter med anden diabetes (se NBV ’Type 1-diabetes’). Dog er regelmæssig undersøgelse for autoimmun sygdom ikke indiceret.
For patienter med GCK-MODY (MODY2) er en årlig diabeteskontrol i et diabetes ambulatorium tilstrækkelig.
Patienter med HNF1B-MODY (MODY5) bør ved diagnosen tilbydes en ultralydsscanning af abdomen mhp. at undersøge for misdannelser af lever og nyre-urinveje. Kvinder som ikke har født bør tilbydes en gynækologisk undersøgelse inklusiv ultralydscanning mhp. at undersøge for misdannelser af genitalia interna. Mænd bør informeres om risiko for nedsat fertilitet (påvirket spermatogenese, aplasi af ductus deferens ses ved HNF1B-MODY) og bør henvises til fertilitetsbehandling ved uhonoreret graviditetsønske hos partner. Patienter med HNF1B-MODY bør monitoreres, fx årligt, med levertal, magnesium og urat i tilslutning til rutine biokemiske analyser (tabel 2), fæces elastase ved mistanke om eksokrin pankreas insufficiens.
Der er rapporteret øget risiko for benigne adenomer i leveren ved patienter med HNF1A-MODY (MODY3) og billeddiagnostiske undersøgelser (UL-scanning af lever) anbefales ved påvirket lever-biokemi og/eller ubehag/trykgener i højre øvre abdominal kvadrant.
Familier med MODY, særligt GCK– eller HNF4A-MODY, informeres om at tilstanden kan kræve særlig opmærksomhed allerede under graviditet (GCK) og neonatalt (HNF4A) både ved bærer-tilstand hos faderen og moderen (se afsnit under MODY og graviditet ovenfor).
Ikke-diabetiske bærere af en kendt sygdomsfremkaldende mutation tilbydes screening for udvikling af diabetes med 2 års interval med start fra 16-års alderen samt før og under graviditet ved hjælp af HbA1c, eventuelt OGTT. Ved behov tilbydes genetisk rådgivning i en klinisk genetisk afdeling.
Referencer
- Shepherd M, Shields B, Hammersley S, Hudson M, McDonald TJ, Colclough K, et al. Systematic Population Screening, Using Biomarkers and Genetic Testing, Identifies 2.5% of the U.K. Pediatric Diabetes Population With Monogenic Diabetes. Diabetes Care. 2016;39(11):1879-88.
- Steele AM, Wensley KJ, Ellard S, Murphy R, Shepherd M, Colclough K, et al. Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: observational case control studies. PLoS One. 2013;8(6):e65326.
- Hattersley AT, Greeley SAW, Polak M, Rubio-Cabezas O, Njølstad PR, Mlynarski W, et al. ISPAD Clinical Practice Consensus Guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2018;19 Suppl 27:47-63.
- Shepherd M, Pearson ER, Houghton J, Salt G, Ellard S, Hattersley AT. No deterioration in glycemic control in HNF-1alpha maturity-onset diabetes of the young following transfer from long-term insulin to sulphonylureas. Diabetes Care. 2003;26(11):3191-2.
- Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362(9392):1275-81.
- Tuomi T, Honkanen EH, Isomaa B, Sarelin L, Groop LC. Improved prandial glucose control with lower risk of hypoglycemia with nateglinide than with glibenclamide in patients with maturity-onset diabetes of the young type 3. Diabetes Care. 2006;29(2):189-94.
- Østoft SH, Bagger JI, Hansen T, Pedersen O, Faber J, Holst JJ, et al. Glucose-lowering effects and low risk of hypoglycemia in patients with maturity-onset diabetes of the young when treated with a GLP-1 receptor agonist: a double-blind, randomized, crossover trial. Diabetes Care. 2014;37(7):1797-805.
- Christensen AS, Hædersdal S, Storgaard H, Rose K, Hansen NL, Holst JJ, et al. GIP and GLP-1 Potentiate Sulfonylurea-Induced Insulin Secretion in Hepatocyte Nuclear Factor 1α Mutation Carriers. Diabetes. 2020;69(9):1989-2002.
- Christensen AS, Hædersdal S, Støy J, Storgaard H, Kampmann U, Forman JL, et al. Efficacy and Safety of Glimepiride With or Without Linagliptin Treatment in Patients With HNF1A Diabetes (Maturity-Onset Diabetes of the Young Type 3): A Randomized, Double-Blinded, Placebo-Controlled, Crossover Trial (GLIMLINA). Diabetes Care. 2020;43(9):2025-33.
- Bellanné-Chantelot C, Chauveau D, Gautier JF, Dubois-Laforgue D, Clauin S, Beaufils S, et al. Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med. 2004;140(7):510-7.
- Spyer G, Macleod KM, Shepherd M, Ellard S, Hattersley AT. Pregnancy outcome in patients with raised blood glucose due to a heterozygous glucokinase gene mutation. Diabet Med. 2009;26(1):14-8.
MIDD
Forekomst
Syndromet Maternally Inherited Diabetes and Deafness (MIDD) omfatter diabetes og varierende grad af bilateral neurosensorisk høretab, som er forårsaget af en mutation i det mitokondrielle DNA (m.DNA).
MIDD forekommer med en hyppighed på 0,8-1,5 % blandt patienter med diabetes. Blandt bærere af m.3243A>G mutationen har omkring 40% diabetes og omkring 70 % vil have udviklet nedsat glukosetolerance eller diabetes, før de er fyldt 70 år (1, 2).
Ætiologi
MIDD forårsages hyppigst af punktmutationen m.3243A>G i det mitokondrielle DNA (maternel arvegang) (3). Mitokondriets produktion af ATP har betydning for glukose-stimuleret insulinsekretion. Nedsat glukose-stimuleret insulinsekretion er en væsentlig årsag til udvikling af diabetes hos patienter med MIDD. Insulinresistens og øget hepatisk glukoneogenese er også observeret (4).
Symptomer og kliniske fund
Klassisk præsentation af MIDD
Patienter med MIDD er karakteriseret ved diabetes, der oftest udvikles i voksenalderen (ofte 30-40 års alderen) og på debuttidspunktet er enten asymptomatisk eller knyttet til symptomer på hyperglykæmi. MIDD debuterer i ca. 8 % af tilfældene med ketoacidose. I de fleste tilfælde vil patienten have haft varierende grader af neurosensorisk høretab forud for udvikling af diabetes. Patienter med MIDD kan desuden præsentere sig med symptomer og kliniske fund, der skyldes en generel påvirkning af mitokondriets funktion. Se tabel 1 for oversigt over de hyppigste kliniske fund og symptomer. Der kan være en familiehistorie med tilsvarende symptomer i den maternelle gren af familien.
Tabel 1. Symptomer og fund hos patienter med MIDD.
| Organsystem | Symptom eller klinisk fund |
| CNS | Epilepsi, migræne, neurosensorisk høretab, retinitis pigmentosa, makula dystrofi, kognitiv påvirkning, forkalkninger i basalganglierne, cerebral apoplexi, herunder occipitale infarkter, atrofi af cerebrum og cerebellum |
| Hjerte | Kardiomyopati, herunder særligt hypertrofi af venstre ventrikel, arytmier, (atrieflimmer, wolff-parkinson white-syndrom, ventrikulære ekstrasystoler) kardiel autonom neuropati |
| Nyre | Nefropati (fokal segmental glomerulosklerose hyppigst, sjældnere diabetisk nefropati). |
| Gastrointestinalt | Obstipation, malabsorption |
| Endokrine kirtler | Diabetes, hypokalkæmi |
| Muskel | Myopati, som primært påvirker muskulaturen i ekstremiteterne (proksimalt), muskelkramper og muskeltræthed. |
Overlappende syndromer
Mutationen m.3243A>G er også knyttet til udvikling af andre syndromer, herunder Myopati, Encefalopati, Laktat-Acidose og Stroke-like episoder (MELAS) samt Kronisk Progressiv Ekstern Oftalmoplegi (CPEO). Patienten med MIDD kan præsentere et eller flere af symptomerne knyttet til disse syndromer og kan også udvikle regelret MELAS (1, 2).
Udredning
Kombinationen af diabetes, høretab og maternel arvegang skal rejse mistanke om MIDD. Patienten har ikke diabetes autoantistoffer (GAD65, ø-celle), niveauet af C-peptid er varierende og lav kropsvægt er hyppig blandt MIDD-patienter. Diagnosen bør understøttes af genetisk screening for m.3243A>G, der kan rekvireres på Molekylærgenetisk Laboratorium, Rigshospitalet (link).
Screening af m.3243A>G
Hos MIDD-patienter indeholder cellerne både normalt DNA og muteret DNA (kaldet heteroplasmi), og forholdet mellem disse former for DNA varierer mellem vævene. Leukocytter har i reglen det laveste niveau af muteret mitokondrielt DNA og heteroplasmigraden i leukocytter falder over tid (5). Fravær af mutation i leukocytter udelukker ikke MIDD; i dé tilfælde skal man bestille screening for mutationen i et andet væv, eksempelvis urin eller mundskrab. Hvis muskelvæv er tilgængeligt, kan dette væv anvendes til screening for mutationen (3).
Tilfælde hvor m.3243A>G ikke kan påvises
Yderligere udredning i disse tilfælde bør varetages i tæt samarbejde med klinisk genetiker, endokrinolog og eventuelt neurolog med særlig interesse for mitokondriesygdom. Hvis screening for øvrige kendte punktmutationer ønskes, anbefales det, at man anvender muskelvæv til genetisk udredning.
Prognose
Diabetes-relaterede komplikationer
Forekomsten af diabetes-relaterede komplikationer er usikker på grund af et begrænset patientantal og det, at mitokondriedysfunktion i sig selv kan medføre påvirkning af blandt andet nyre og nervevæv. Prævalensen af perifer neuropati er omkring 50 % ved MIDD. Ca. 20 % oplever gastrointestinal dysmotilitet, der kan forveksles med gastroparese. Forekomsten af diabetisk retinopati er ikke afklaret. Ikke-diabetisk nefropati er hyppigt forekommende, og proteinuri skal som udgangspunkt opfattes som værende relateret til anden nyresygdom end diabetisk nefropati (6).
Behandling
Kolesterolreducerende behandling: Ingen evidens foreligger vedrørende kolesterolreducerende behandling. Statiner påvirker mitokondriefunktionen medførende nedsat ATP produktion. Dette er en medvirkende årsag til statin-induceret myopati (7). Statiner kan således øge risikoen for myopati hos patienter med mitokondriesygdom. Der er ikke enighed om brugen af statiner til patienter med MIDD. I et review fra Ramachandran et al. anbefales at statiner undgås, specielt hos patienter med myopati (8). Shaefer et al. er mindre restriktiv, men anbefaler forsigtighed samt måling af CK før start af behandling samt gentage CK målinger under behandling (9). Endelig skal patienterne rapportere hvis der opstår nye myopatigener. Ved behandlingsbehov kan ezetimib eller et fibrat anvendes.
Blodtrykssænkende behandling: Behandling af hypertension følger guidelines for type 2-diabetes.
Glukosereducerende behandling: Mål for glykæmisk kontrol følger almindelige rekommandationer (link NBV T2DM). Initialt afklares, om patienten er insulinopen, idet op mod 13-17 % af MIDD-patienterne har insulinbehov på diagnosetidspunktet. Glykæmisk kontrol vil hos mange patienter kunne opnås ved diæt, livsstilsændringer og/eller peroral antidiabetisk behandling. Over en periode på 2-4 år vil de fleste patienter udvikle behov for insulinbehandling (10).
Valg af behandling
Ingen kliniske studier støtter, at man bør foretrække en type peroral antidiabetisk lægemiddel eller én type insulin frem for en anden (9).
SU-præparater foretrækkes hos yngre patienter, da patienterne har tendens til lav kropsvægt og i reglen reagerer på stimulering af insulinsekretionen. Brug af metformin er ikke undersøgt i patienter med MIDD, men der er generelt enighed om at metformin bør undgås på grund af potentielt øget risiko for laktatacidose. Hvis metformin bruges, bør laktatniveauet kontrolleres løbende, og patienten skal instrueres i at stoppe med behandlingen ved svære infektioner og anden interkurrent sygdom. DPP-IV-hæmmere og GLP1-analoger har været brugt hos MIDD-patienter (9). Der er ingen erfaring med behandling med SGLT-2i. Insulinbehandling følger vanlige guidelines for opstart af insulinbehandling (link T2DM NBV).
Medicin, som bør undgås
Lægemidler, som påvirker mitokondriets funktion og medfører risiko for påvirkning af hørelse, bør undgås, herunder tetracycliner, valproat, fenytoin.
Graviditet
Risikoen for udvikling af diabetes under graviditeten er forøget hos bærere af m.3243A>G (ca. 11 %), og risikoen for præeklampsi og præmatur fødsel er forøget (11). Ikke-diabetiske gravide mutationsbærere og gravide med en betydelig familieanamnese med diabetes og mitokondriesygdom bør screenes for gestationel diabetes med en oral glukosebelastning.
Gravide med MIDD behandles med insulin i graviditeten. For yderligere information om diabetesbehandlingen i graviditeten med insulin og kostvejledning henvises til NBV ’Diabetes og Graviditet’: https://endocrinology.dk/index.php/1-diabetes-mellitus/8-diabetes-og-graviditet
Visitation og opfølgning
Som udgangspunkt bør MIDD-patienter følges hos endokrinolog med særlig interesse for sygdommen og med adgang til multidiciplinær vurdering og behandling på tværs af specialer.
Det anbefales, at der foretages en klinisk vurdering af udvikling af neurologisk og kardiologisk sygdom hvert år. Screening for klassiske diabeteskomplikationer inklusiv neuro-, nefro- og retinopati skal ske årligt.
Kardiologisk vurdering bør overvejes ved symptomer på hjertesvigt eller arytmier, og ekkokardiografi anbefales ved det fyldte 35. år og efterfølgende hvert 3-5 år (3). Nefrologisk vurdering anbefales ved makroalbuminuri. Vurdering af hørelsen bør foretages ved diagnosen og herefter regelmæssigt, da en stor andel vil være kandidater til og får behov for cochlear implantat. Endvidere bør der foretages radiologisk udredning på vid indikation, hvis der udvikles neurologiske symptomer.
Fertile kvinder der er bærere af m.3243A>G mutationen bør henvises til klinisk genetisk afdeling med henblik på prækonceptionel rådgivning.
Det anbefales, at ikke-diabetiske mutationsbærere screenes for diabetes med måling af HbA1c hvert år. For yderligere information anbefales information på Newcastle Mitochondrial Centre, NHS Specialised Services for Rare Mitochondrial Disorders of Adults and Children’s hjemmeside (http://www.newcastle-mitochondria.com/service/patient-care-guidelines/).
Referencer
- Nesbitt V, Pitceathly RD, Turnbull DM, Taylor RW, Sweeney MG, Mudanohwo EE, et al. The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m.3243A>G mutation–implications for diagnosis and management. J Neurol Neurosurg Psychiatry. 2013;84(8):936-8.
- Maassen JA, LM TH, Van Essen E, Heine RJ, Nijpels G, Jahangir Tafrechi RS, et al. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;53 Suppl 1:S103-9.
- Murphy R, Turnbull DM, Walker M, Hattersley AT. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet Med. 2008;25(4):383-99.
- de Andrade PB, Rubi B, Frigerio F, van den Ouweland JM, Maassen JA, Maechler P. Diabetes-associated mitochondrial DNA mutation A3243G impairs cellular metabolic pathways necessary for beta cell function. Diabetologia. 2006;49(8):1816-26.
- Langdahl JH, Larsen M, Frost M, Andersen PH, Yderstraede KB, Vissing J, et al. Lecocytes mutation load declines with age in carriers of the m.3243A>G mutation: A 10-year Prospective Cohort. Clin Genet. 2018;93(4):925-8.
- Seidowsky A, Hoffmann M, Glowacki F, Dhaenens CM, Devaux JP, de Sainte Foy CL, et al. Renal involvement in MELAS syndrome – a series of 5 cases and review of the literature. Clin Nephrol. 2013;80(6):456-63.
- Balestrino M, Adriano E. Creatine as a Candidate to Prevent Statin Myopathy. Biomolecules. 2019;9(9).
- Ramachandran R, Wierzbicki AS. Statins, Muscle Disease and Mitochondria. J Clin Med. 2017;6(8).
- Schaefer AM, Walker M, Turnbull DM, Taylor RW. Endocrine disorders in mitochondrial disease. Mol Cell Endocrinol. 2013;379(1-2):2-11.
- Guillausseau PJ, Massin P, Dubois-LaForgue D, Timsit J, Virally M, Gin H, et al. Maternally inherited diabetes and deafness: a multicenter study. Ann Intern Med. 2001;134(9 Pt 1):721-8.
- de Laat P, Fleuren LH, Bekker MN, Smeitink JA, Janssen MC. Obstetric complications in carriers of the m.3243A>G mutation, a retrospective cohort study on maternal and fetal outcome. Mitochondrion. 2015;25:98-103.
NDM
Forekomst
Neonatal diabetes er sjælden med en incidens mellem 1:90.000 og 1:215.000 nyfødte (1, 2). Definitionen er vanligvis hyperglykæmi debuterende i de første 6 måneder af livet, der persisterer mere end 14 dage og er behandlingskrævende.
Ætiologi
Den genetiske ætiologi til NDM er heterogen med mere end 20 forskellige gener associeret med isoleret NDM samt syndromiske former for NDM. Blandt patienter diagnosticeret med diabetes før 6-måneders alderen er en polygen, autoimmun genese til betacellesvigtet særdeles sjælden, og ved genetisk udredning kan man påvise en sygdomsfremkaldende mutation i mere end 80 % af patienterne (tabel 1)(3).
Tabel 1. Prævalens (%) af hyppige genetiske ætiologier til NDM
| Gen/locus | TNDM | PNDM | Original reference |
| 6q24 | 70 | – | https://www.ncbi.nlm.nih.gov/pubmed/10699182 |
| KCNJ11 | 12 (AD*) | 30 (AD) | https://www.ncbi.nlm.nih.gov/pubmed/15115830 |
| ABCC8 | 13 (AD) | 14 (AD) | https://www.ncbi.nlm.nih.gov/pubmed/16885549 |
| INS | <1 (AR*) | 10 (AD/AR) | https://www.ncbi.nlm.nih.gov/pubmed/17855560 |
| GCK | – | 1 (AR) | https://www.ncbi.nlm.nih.gov/pubmed/14578306 |
*AD – Autosomal dominant; AR – Autosomal recessivt
Symptomer og kliniske fund
NDM defineres ved diabetes, som diagnosticeres inden for barnets første halve til hele leveår.
- 20 % af patienterne har transient NDM, hvor patienterne har behandlingsbehov i de første levemåneder, men hvor blodglukose spontant normaliseres inden for barnets første leveår. Omkring 50 % af patienterne udvikler diabetes igen omkring pubertetens indtræden
- De resterende 80 % af patienterne har permanent NDM og har et livslangt behandlingsbehov
NDM præsenterer sig på flere måder:
- Tilfældig opsporing af hyperglykæmi hos prætermt, præmaturt og/eller intrauterint væksthæmmet barn (IUGR) i forbindelse med intensiv overvågning i de første levedøgn
- Barnet bliver akut dårligt, eventuelt med bevidsthedssløring på grund af diabetisk ketoacidose (>60 % af ikke-hospitaliserede børn med hidtil uerkendt NDM debuterer med diabetisk ketoacidose)
- Symptomgivende hyperglykæmi (polydipsi, polyuri, trivselsproblemer)
- Asymptomatisk hvor forhøjet blodglukose påvises ifm. analyse af andre blodprøver
Nyfødte kan have forbigående forhøjet blodglukose på grund af infektion, fysiologisk stress, parenteral tilførsel af glukose, steroidbehandling, præterm fødsel (< 28 uge), IUGR (især < 1500 g) eller præmaturitet uden at have regelret diabetes. Prævalensen af hyperglykæmi hos præterme nyfødte er mellem 25-75 % med debut i løbet af de første 10 levedøgn og normalisering efter 2-3 døgn. Følgende kan overvejes ift. at differentiere mellem regelret diabetes og forbigående hyperglykæmi af anden årsag:
- Overvej diagnosen ved intermitterende hyperglykæmi (BG>11 mmol/L) varende mere end 14 dage, hvor der ikke er en alternativ forklaring
- Overvej diagnosen ved alle børn < 6-12 måneder ved BG over 15 mmol/L uanset varighed, barnets alder og almen tilstand
- Overvej diagnosen hos ethvert barn som har behov for insulinbehandling før 6-12 måneders alderen
Kliniske fund ved diabetesdebut kan omfatte:
- Lav kropsvægt, tegn på dehydratio (nedsat hud turgor, indsunken fontanelle, hallonerede øjne, påvirket kredsløb)
- Påvirket bevidsthed, keton foetor, takypnø (Kussmauls respiration)
- Specifikke fund som kan relateres til subtype af NDM (tabel 2)
Tabel 2. Kliniske karakteristika hos patienter med NDM
| Gen/locus | Patofysiologi | Fænotype | Øvrige problemstillinger | Behandling |
| KCNJ11/ABCC8 | Defekt i betacelle ATP-følsomme kaliumkanal | TNDM eller PNDM <6-9 måneders alderen, IUGR, ofte DKA ved debut Risiko for komplikationer som ved T1, dog manglende viden ved TNDM | Varierende sværhedsgrad af udviklingsforstyrrelser, epilepsi (syndromerne iDEND og DEND), ADHD, søvnforstyrrelser | SU Insulin |
| INS | AD: Betacelle stress AR: Ophørt/nedsat ekspression af INS genet | PNDM <12 måneders alderen, let IUGR, ofte DKA ved debut (AD). PNDM i første leveuge, i sjældne tilfælde TNDM, svær IUGR (AR) Risiko for komplikationer som ved T1 | Tidlig katarakt | Insulin |
| 6q24 | Paternel overekspression af 6q24, nedsat insulin sekretion, normal IS | TNDM i første levedøgn/ uger, svær IUGR, DKA sjælden, diabetes remission i løbet af første leveår, relaps hos 50 % i puberteten Ukendt risiko for komplikationer | Makroglossi, umbilikal hernie | Insulin SU Observation |
| GCK (AR) | Manglende GSIS | PNDM med debut i første levedøgn/uger, svær IUGR, DKA sjælden Risiko for komplikationer forventes at være som ved T1 | GCK-MODY (MODY2) hos evt. børn | Insulin |
AD Autosomal dominant; Autosomal recessiv; DEND Developmental delay, epilepsi, neontal diabetes; DKA Diabetisk ketoacidose; GSIS Glukose stimuleret insulinsekretion; iDEND Intermediate Developmental delay, epilepsi, neontal diabetes; IS Insulinfølsomhed; IUGR Intrauterin væksthæmning; PNDM Permanent neonatal diabetes; SU Sulfonylurinstof; TNDM Transient neonatal diabetes
Udredning
Syre-base status, ketoner i blod og urin, C-peptid og evt. GAD65 antistof og/eller ø-celle antistof.
Ultralyd scanning af abdomen med henblik på at visualisere pankreas.
Alle børn med transient eller permanent diabetes med debut før 6 måneders alderen bør tilbydes genetisk udredning. Børn diagnosticeret med diabetes fra 6 til 12 måneders alderen og som ikke har tegn på pankreatisk autoimmunitet skal også tilbydes genetisk udredning. Detaljer om rekvisition af genetisk udredning er beskrevet i særskilt afsnit om MODY (link). Undersøgelse af genetiske defekter omkring 6q24 kan rekvireres på Kennedy Centret, København (http://www.kennedy.dk/).
Prognose
Der foreligger sparsom viden, men risikoen for sendiabetiske komplikationer forventes at være som ved type 1-diabetes ved PNDM, hvorimod risikoen ved TNDM er ukendt. Ligesom i anden diabetesbehandling er god glykæmisk kontrol og reduktion af øvrige kardiovaskulære risikofaktorer essentiel for en god prognose. Effekten af SU ved KCNJ11/ABCC8 NDM (se afsnit om behandling) på sendiabetiske komplikationer kendes ikke.
Behandling
Kolesterolreducerende behandling: Ingen evidens foreligger vedrørende kolesterolreducerende behandling. Der anbefales en tilgang som ved type 1-diabetes (link).
Blodtrykssænkende behandling: Ingen studier foreligger vedrørende blodtrykssænkende behandling, men behandlingsmål som ved type 1-diabetes anbefales.
Glukosereducerende behandling: Ingen langtidsstudier viser evidens for glukosereducerende behandling, men der bør stiles mod glykæmisk kontrol svarende til anbefalinger for patienter med type 1-diabetes.
Patienter med en mutation i KCNJ11 eller ABCC8
Op mod 90 % af patienter med en mutation i KCNJ11 responderer på behandlingen med høj-dosis SU (0.5-1.0 mg/kg/døgn) og kan varetages uden insulin-behandling (4, 5). Success-raten er lavere for patienter med en mutation i ABCC8, men en stor del af patienterne responderer helt eller delvist. SU-behandlingen er mere effektiv end insulin-behandling i forhold til glykæmisk regulation, behandlingseffekten er vedvarende, og behandlingen er ikke forbundet med betydelig hypoglykæmi. SU-behandlingen kan bedre de eventuelle mutations specifikke neuro-kognitive deficits. Tidlig opstart af SU-behandling er måske vigtig for at opnå et optimalt glykæmisk behandlingsrespons og måske også i forhold til patientens neuro-kognitive udvikling. For praktiske deltaljer omkring opstart af SU-behandling eller eventuelt skifte fra insulin til SU-behandling henvises til: https://www.diabetesgenes.org/about-neonatal-diabetes/ (6-8).
Øvrige former for neonatal diabetes
Patienter med 6q24-TNDM behandles med insulin ved behandlingsbehov, men enkelte kasuistikker har beskrevet respons på SU-behandling (9). Der er manglende erfaring omkring behandlingen ved eventuelt recidiv af diabetes omkring puberteten, men patienterne har endogen insulinproduktion og tabletbehandling kan være tilstrækkelig.
Øvrige patienter med NDM behandles med insulin ved behandlingsbehov.
Behandling af NDM under graviditet
I Danmark anbefales behandling med insulin under graviditet hos voksne patienter med PNDM og hos voksne patienter med TNDM som udvikler diabetes efter neonatal perioden. For yderligere information om diabetesbehandlingen i graviditeten med insulin og kostvejledning henvises til NBV ’Diabetes og Graviditet’. Der er manglende viden om, hvordan personer med TNDM, som ikke har udviklet diabetes i voksenlivet, håndteres i forbindelse med graviditet. Der anbefales hos disse kvinder undersøgelse for diabetes forud for en planlagt graviditet med Hb1Ac. OGTT anbefales ved glukosuri (urinstix) ved svangrekontroller samt uge 14-20 og gentaget ved uge 27-30 såfremt OGTT i uge 14-20 var normal.
Børn af patienter med AD NDM (mutationer i KCNJ11, ABCC8 eller INS) har 50 % risiko for at have arvet mutationen og dermed NDM. Ved IUGR hos et barn med NDM i familien er der bestyrket mistanke og blodglukose skal monitoreres efter fødslen. Genetisk undersøgelse bør tilbydes kort efter fødslen. Børn af patienter med NDM pga. homozygot GCK-mutation, har GCK-MODY (MODY2). Børn af patienter med en 6q24 abnormitet har en variabel risiko for at have arvet den genetiske defekt fra omkring 0 % til 50 % afhængig af typen af genetisk defekt og om barnets far eller mor bærer den genetiske defekt. Hos disse patienter anbefales genetisk rådgivning forud for graviditet.
Visitation og opfølgning
Patienter med PNDM har risiko for udvikling af sendiabetiske komplikationer og bør følge diabeteskontroller som beskrevet for type 1-diabetes og MODY1, 3 og 5. I barnets første leveår er behandlingen kompleks og tæt kontakt til et diabetesambulatorium er nødvendig. Patienter med TNDM følges tæt i det første leveår med henblik på at nedjustere behandlingen i takt med den gradvise normalisering af blodglukose. Blandt de 50 % af patienterne med TNDM, som udvikler diabetes i pubertetsalderen, oplever mange i den mellemliggende periode forbigående, selvlimitterende hyperglykæmi i tilslutning til interkurrent sygdom, ligesom en del patienter, af ukendt årsag, oplever episoder med hypoglykæmi (10). Patienter med TNDM, som er ovre den behandlingskrævende fase af deres sygdom, kan tilbydes en årlig kontrol i et diabetes ambulatorium med henblik på at sikre rettidig diagnosticering af stigende blodglukose og eventuelt hjælp til håndtering af hypo/hyperglykæmiske episoder.
Genetisk rådgivning i en klinisk genetisk afdeling kan overvejes ved behov.
Referencer
- Iafusco D, Massa O, Pasquino B, Colombo C, Iughetti L, Bizzarri C, et al. Minimal incidence of neonatal/infancy onset diabetes in Italy is 1:90,000 live births. Acta Diabetol. 2012;49(5):405-8.
- Stanik J, Gasperikova D, Paskova M, Barak L, Javorkova J, Jancova E, et al. Prevalence of permanent neonatal diabetes in Slovakia and successful replacement of insulin with sulfonylurea therapy in KCNJ11 and ABCC8 mutation carriers. J Clin Endocrinol Metab. 2007;92(4):1276-82.
- Lemelman MB, Letourneau L, Greeley SAW. Neonatal Diabetes Mellitus: An Update on Diagnosis and Management. Clin Perinatol. 2018;45(1):41-59.
- Bowman P, Mathews F, Barbetti F, Shepherd MH, Sanchez J, Piccini B, et al. Long-term Follow-up of Glycemic and Neurological Outcomes in an International Series of Patients With Sulfonylurea-Treated ABCC8 Permanent Neonatal Diabetes. Diabetes Care. 2021;44(1):35-42.
- Bowman P, Sulen Å, Barbetti F, Beltrand J, Svalastoga P, Codner E, et al. Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: an international cohort study. Lancet Diabetes Endocrinol. 2018;6(8):637-46.
- Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, Larkin B, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med. 2006;355(5):467-77.
- Rafiq M, Flanagan SE, Patch AM, Shields BM, Ellard S, Hattersley AT. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care. 2008;31(2):204-9.
- Babiker T, Vedovato N, Patel K, Thomas N, Finn R, Männikkö R, et al. Successful transfer to sulfonylureas in KCNJ11 neonatal diabetes is determined by the mutation and duration of diabetes. Diabetologia. 2016;59(6):1162-6.
- Carmody D, Beca FA, Bell CD, Hwang JL, Dickens JT, Devine NA, et al. Role of noninsulin therapies alone or in combination in chromosome 6q24-related transient neonatal diabetes: sulfonylurea improves but does not always normalize insulin secretion. Diabetes Care. 2015;38(6):e86-7.
- Flanagan SE, Mackay DJ, Greeley SA, McDonald TJ, Mericq V, Hassing J, et al. Hypoglycaemia following diabetes remission in patients with 6q24 methylation defects: expanding the clinical phenotype. Diabetologia. 2013;56(1):218-21.
Tovholder
Oprettet: Oktober 2018
1. Revision: Oktober 2021
Næste revision: Oktober 2024
Øvrig arbejdsgruppe:
Herborg Johannesen
Anette Prior Gjesing
Henrik Maagensen
Jesper Krogh
Torben Hansen
Tina Vilsbøll
Rikke Vendelbo Viggers
Ulla Kampmann Opstrup
Per Heden Andersen
Bidrag af:
Kurt Kristensen (pædiatri)
Lene Ringholm (endokrinologi)
Interessekonflikter